








































 chemistry
chemistrySimilar presentations:

")

")



")
 методы определения концентрации белка")

Общая характеристика физико-химических методов анализа лекарственных веществ
1. Лекция № 8-9
«Общая характеристика физикохимических методов анализалекарственных веществ.»
1
2. Основные приемы, используемые в физико-химических методах анализа
Почти во всех физико-химических методах анализа применяются дваосновных методических приема: метод прямых измерений и метод
титрования (метод косвенных измерений).
Прямые методы. В этих методах используется зависимость
аналитического сигнала от природы анализируемого вещества и его
концентрации. Свойством, зависящем от природы вещества, является,
например, длина волны спектральной линии в эмиссионной
спектроскопии, потенциал полуволны в полярографии, а количественной
характеристикой служит интенсивность сигнала - интенсивность
спектральной линии в первом случае, сила диффузионного тока — во
втором. В некоторых методах связь аналитического сигнала с природой
вещества установлена строго теоретически. Например, линии в спектре
атома водорода могут быть рассчитаны по теоретически выведенным
формулам с использованием фундаментальных констант (постоянная
Планка, заряд электрона и т. д.).
2
3.
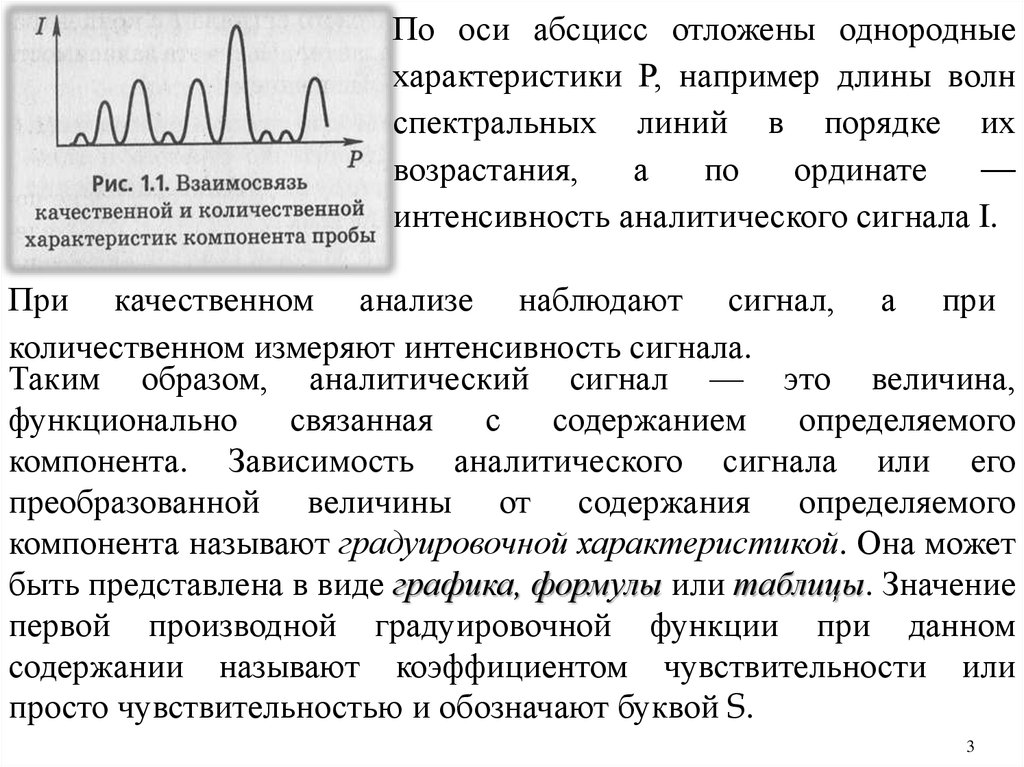
По оси абсцисс отложены однородныехарактеристики Р, например длины волн
спектральных линий в порядке их
возрастания,
а
по
ординате
—
интенсивность аналитического сигнала I.
При качественном анализе наблюдают сигнал, а при
количественном измеряют интенсивность сигнала.
Таким образом, аналитический сигнал — это величина,
функционально
связанная
с
содержанием
определяемого
компонента. Зависимость аналитического сигнала или его
преобразованной величины от содержания определяемого
компонента называют градуировочной характеристикой. Она может
быть представлена в виде графика, формулы или таблицы. Значение
первой производной градуировочной функции при данном
содержании называют коэффициентом чувствительности или
просто чувствительностью и обозначают буквой S.
3
4.
Область значений определяемых содержаний, предусмотреннаяданной методикой, составляет диапазон определяемых содержаний.
Наименьшее значение определяемого содержания называют нижней
границей определяемых концентраций или нижним пределом сн.
Наибольшее значение — верхней границей или верхним пределом св.
Наименьшее содержание, при котором по данной методике можно
обнаружить присутствие определяемого компонента с заданной
доверительной вероятностью Р (обычно Р=0,95), называют пределом
обнаружения и обозначают cmin,P (например, cmin,P = 0,95). Предел
обнаружения находят по величине минимально обнаруживаемого
аналитического сигнала.
4
5.
Связь интенсивности аналитического сигнала I с концентрациейвещества имеет различный характер. Часто эта зависимость
выражается простым линейным соотношением:
I =Ас ,
(1.1)
где А — константа; с — концентрация.
В аналитической практике наибольшее распространение
получили
следующие
методы
прямого
количественного
определения с помощью физико-химических измерений: 1) метод
градуировочного графика; 2) метод молярного свойства; 3) метод
добавок. Все они основаны на использовании стандартных образцов
или стандартных растворов.
5
6. Метод градуировочного графика
В этом методе измеряется интенсивность аналитического сигналаI у нескольких стандартных образцов или нескольких стандартных
растворов и строится градуировочный график обычно в
координатах I = f(с), где с — концентрация определяемого
компонента в стандартном образце или стандартном растворе. Затем
в тех же условиях измеряется интенсивность сигнала у
анализируемой пробы и по градуировочному графику находится
концентрация анализируемого вещества. Интервал концентраций на
градуировочном графике должен охватывать предполагаемую
область анализируемых концентраций, а состав стандартного
образца или раствора должен быть близок к составу
анализируемого.
6
7. Метод молярного свойства
Измеряется интенсивность аналитического сигнала у несколькихстандартных образцов или растворов и рассчитывается молярное
свойство А, т.е. интенсивность аналитического сигнала,
пропорциональная 1 моль вещества: А = I/сст. Затем в тех же
условиях измеряется интенсивность сигнала у анализируемой
пробы и по соотношению сх = I/A рассчитывается концентрация
анализируемого компонента. Метод предполагает строгое
соблюдение соотношения (1.1), по крайней мере, в области
анализируемых концентраций.
7
8. Метод добавок
В этом методе сначала измеряется интенсивность аналитического сигналапробы, затем в пробу вводится известный объем стандартного раствора до
концентрации сст и снова измеряется интенсивность сигнала. Если Ixинтенсивность аналитического сигнала пробы, а Ix + ст — интенсивность сигнала
после добавки стандартного раствора, то, очевидно,
откуда
(1.2)
Метод также предполагает строгое соблюдение соотношения (1.1). Уравнение
(1.2) нередко решается графически.
8
9.
Методы титрования. В этих методах в ходе титрованияизмеряется интенсивность аналитического сигнала I и строится
кривая титрования в координатах I-V, где V — объем добавленного
титранта, мл. Точка эквивалентности находится по кривой
титрования. Виды кривых титрования весьма многообразны, так как
интенсивность аналитического сигнала может быть связана с
концентрацией определяемого вещества, титранта или продукта
реакции.
9
10. Лекция № 8-9
«Определение физико-химическихсвойств субстанций и растворов ЛВ.»
10
11. Определение степени белизны порошкообразных лекарственных средств
Физический метод, впервые включенный в ГФ XI. Степень белизны(оттенка) твердых лекарственных веществ можно оценить различными
инструментальными методами на основе спектральной характеристики
света, отраженного от образца. Для этого измеряют коэффициенты
отражения при освещении образца белым светом, полученным от
специального источника со спектральным распределением или
пропущенным через светофильтры с максимумом пропускания 614 нм
(красный) или 459 нм (синий). Можно также измерять коэффициент
отражения света, пропущенного через зеленый светофильтр (522 нм).
Коэффициент отражения — это отношение величины отраженного
светового потока к величине падающего светового потока. Для белых или
белых с сероватым оттенком веществ степени белизны теоретически равна
1. Вещества, у которых она 0,95—1, а степени яркости < 0,85, имеют
сероватый оттенок.
11
12.
Более точно оценку белизны лекарственных веществ можноосуществить с помощью спектрофотометров отражения, например
СФ-18, выпускаемых ЛОМО (Ленинградским оптико-механическим
объединением). Интенсивность цветовых или сероватого оттенков
устанавливают по абсолютным коэффициентам отражения.
Значения степени белизны и степени яркости являются
характеристиками качества белых и белых с оттенками
лекарственных веществ. Их допустимые пределы регламентируются
в частных статьях.
12
13. Определение температуры плавления
Это температура, при которой твердое тело находится вравновесии с жидкой фазой при насыщенной фазе пара.
Под температурой плавления вещества подразумевают интервал
температуры между началом плавления — появлением первой
капли жидкости и концом плавления — полным переходом
вещества в жидкое состояние.
Приведенные в частных статьях фармакопеи интервалы
температур плавления указывают на то, что наблюдаемая
температура плавления данного препарата должна находиться в
указанных пределах, при этом интервал между началом и концом
плавления не должен превышать 2° С. Отдельные отклонения от
этого интервала должны быть указаны в частных статьях.
13
14.
В случаях нечеткого начала или конца плавления отдельныхпрепаратов вместо интервала температуры плавления можно
определять только конец плавления или только начало плавления.
Тогда приведенный в частных статьях интервал температуры
плавления указывает, что начало плавления (или конец плавления)
должно укладываться в этих пределах.
Для веществ, неустойчивых при нагревании, определяют
температуру разложения. Температурой разложения называют
температуру, при которой происходит резкое изменение веществ
(вспенивание).
В зависимости от физических свойств веществ следует применять
один из нижеприведенных методов определения температуры
плавления.
Методы 1 и 1а — для твердых веществ, легко превращаемых в
порошок: устойчивых при нагревании (метод 1) и неустойчивых при
нагревании (метод 1а).
Методы 2 и 3 — для веществ, не растирающихся в порошок, как
жиры, воск, парафин, вазелин, смолы.
14
15.
Для определения температуры плавления по методам 1, 1а и 2 допускаютсядва прибора.
I. «Прибор для определения температуры плавления с диапазоном измерений
в пределах от 20 до 360°С» (ПТП) с электрическим обогревом. Прибор состоит
из следующих частей:
основание со щитком управления и номограммой;
стеклянный
блок-нагреватель,
обогрев
которого
осуществляется
константановой проволокой, навитой бифилярно;
оптическое приспособление (ГОСТ 7594—75);
приспособление для установки термометров;
приспособление для установки капилляров;
термометр укороченный с ценой деления 0,5° С;
источник нагрева (электрический обогрев);
капилляры длиной 20 см.
15
16.
II. Второй прибор состоит из следующих частей:круглодонная колба из термостойкого стекла вместимостью от 100 до 150 мл;
длина горла колбы 20 см; диаметр горла от 3 до 4 см;
пробирка из термостойкого стекла, вставленная в колбу и отстоящая от дна
колбы на расстоянии 1 см; диаметр пробирки от 2 до 2,5 см;
термометр ртутный стеклянный укороченный с ценой деления 0,5° С;
источник нагрева (газовая горелка, электрический обогрев);
капилляры.
Колбу наполняют на 3/4 объема шара соответствующей жидкостью;
вазелиновое масло (ГОСТ 3164—78) или жидкие силиконы;
концентрированная серная кислота (ГОСТ 4204—77) — для веществ с
температурой плавления от 80 до 260°С;
раствор 3 частей калия сульфата (ГОСТ 4145—74) в 7 частях (массовых)
концентрированной серной кислоты (ГОСТ 4204—77) — для веществ с
температурой плавления выше 260°С;
дистиллированная вода — для веществ с температурой плавления ниже 80°С.
16
17. Методика определения
Метод 1 и 1а. Если в частных статьях нет других указаний, тонкоизмельченное вещество сушат при температуре от 100 до 105°С в течение 21ч или
в эксикаторе над серной кислотой в течение 24 ч. Любые другие условия должны
быть указаны в частных статьях. Высушенное вещество помещают в капилляр,
имеющий диаметр от 0,9 до 1 мм и толщину стенки от 0,1 до 0,15 мм, запаянный
с одного конца.
При плавлении в приборе ПТП длина капилляра должна быть 20 см, в случае
второго прибора — от 6 до 8 см. Для уплотнения вещества капилляр многократно
бросают в стеклянную трубку высотой не менее 50 см, поставленную
вертикально на стекло. Высота слоя вещества в капилляре должна быть около 3
мм. Капилляр с веществом сохраняют до начала определения в эксикаторе.
Во внутреннюю пробирку второго прибора помещают термометр так, чтобы
конец его отстоял от дна пробирки на 1 см.
Нагревание в обоих приборах проводят сначала быстро, а затем регулируют
его так, чтобы за 10°С до начала плавления была достигнута необходимая
скорость подъема температуры, указанная ниже.
17
18.
За 10°С до ожидаемого начала плавления капилляр с веществом вносят вприборы (первый или второй) таким образом, чтобы запаянный конец его
находился на нижней части столика, расположенной на уровне середины ртутного
шарика термометра. При плавлении во втором приборе капилляр должен быть
расположен таким образом, чтобы запаянный его конец находился на середине
ртутного шарика термометра.
Продолжают нагревание со скоростью:
для веществ, плавящихся по методу 1, при определении температуры
плавления ниже 100°С — со скоростью от 0,5 до 1°С в 1 мин; при определении
температуры плавления от 100 до 150°С — от 1 до 1,5°С в 1 мин; при
определении температуры плавления выше 150°С — от 1,5 до 2°С в 1 мин;
для веществ, плавящихся по методу 1а, — от 2,5 до 3,5°С в 1 мин.
Проводят не менее двух определений; за температуру плавления принимают
среднее арифметическое значение нескольких определений, проведенных в
одинаковых условиях и отличающихся друг от друга не более чем на 1°С.
В случае расхождений при определении температуры плавления на разных
приборах в частной статье должна быть приведена температура плавления на
каждом приборе.
18
19.
Метод 2. а) Для мягких веществ: капилляр длиной 20 см при применениипервого прибора и от 6 до 8 см — при применении второго прибора и внутренним
диаметром от 1 до 2 мм, открытый с обоих концов, погружают в вещество так,
чтобы оно заполнило нижнюю часть капилляра и образовало слой высотой около
10 мм.
б) Для твердых веществ: испытуемое вещество расплавляют на бане при
возможно более низкой температуре, тщательно перемешивают, набирают его в
капилляр, как указано выше (см. метод 2а), и оставляют при температуре 0°С в
течение от 1 до 2 ч.
С заполненным тем или другим способом капилляром проводят определение
температуры плавления по методу 1.
За температуру плавления принимают ту температуру, при которой столбик
вещества становится жидким, поднимаясь в некоторых случаях по капилляру.
Проводят не менее двух определений. За температуру плавления принимают
среднее значение. Расхождение между двумя определениями не должно
превышать 1°С.
19
20.
Метод 3. При этом методе применяют термометр типа Убеллоде (ГОСТ400—80 Е). Определение проводят следующим образом. Чашечку 1 (рис. 1)
заполняют исследуемым веществом, избегая по возможности попадания
пузырьков воздуха, и вставляют ее в нижнюю часть гильзы 2 до упора. Ртутный
шарик термометра 3 при этом погружается в вещество, излишек которого
выдавливается через боковые отверстия 4 гильзы. Последнюю тщательно
протирают и термометр помещают в пробирку 5 длиной от 19 до 21 см и
диаметром от 4 до 4,5 см с помощью пробки с прорезом таким образом, чтобы
нижняя часть чашечки отстояла от дна пробирки на 2,5 см. Пробирку укрепляют в
вертикальном положении в стакане 6 так, чтобы она была погружена на 2/3 в воду
и нижний ее конец при этом отстоял от дна стакана на 2,5 см. Начинают нагревать
прибор при постоянном перемешивании жидкости с помощью мешалки 7. Когда
температура будет на 15-20°С ниже ожидаемой, регулируют нагревание таким
образом, чтобы температура поднималась на 1°С в 1 мин. За температуру
плавления принимают температуру, при которой из отверстия 8 упадет первая
капля расплавленного вещества.
Проводят не менее двух определений; за температуру плавления принимают
среднее значение. Расхождение между двумя определениями не должно
превышать 1°С.
20
21.
2122. Определение температуры затвердевания
Температурой затвердевания называют наиболее высокую, остающуюся втечение короткого времени постоянной температуру во время перехода вещества
из жидкого состояния в твердое.
Определение проводят в приборе (рис. 2), состоящем из толстостенной
пробирки 1 с внутренним диаметром 20±1 мм, снабженной пробкой, в которой
укреплены термометр 2 и мешалка 3. Рекомендуются укороченные термометры с
ценой деления шкалы 0,5°С (ГОСТ 215—73Е). Мешалку можно применять
стеклянную или из проволоки, согнутую на конце петлей под прямым углом.
Пробирку укрепляют на пробке во второй
толстостенной наружной пробирке 4 (диаметром около 35
мм), служащей воздушной баней. Прибор помещают в
сосуд 5 вместимостью 1000 мл, наполняемый водой или
охладительной смесью таким образом, чтобы уровень
жидкости в сосуде был выше вещества во внутренней
пробирке. Температуру в сосуде измеряют с помощью
второго термометра 6.
Вместо указанных выше пробирок (1,4) можно
использовать прибор Жукова (ГОСТ 4255—75).
22
23.
Методика определения. 10 г испытуемого вещества, находящегося в жидкомсостоянии (твердое вещество предварительно расплавляют при возможно более
низкой температуре), помещают во внутреннюю сухую пробирку прибора и
укрепляют термометр таким образом, чтобы ртутный шарик находился
посередине слоя испытуемого вещества.
Пробирку с веществом вставляют в наружную пробирку и укрепляют в
сосуде, жидкость в котором должна иметь температуру на 5°С ниже ожидаемой
температуры затвердевания.
При постоянном перемешивании испытуемого вещества отмечают
температуру каждые 30с. Вначале происходит постепенное понижение
температуры, затем, при появлении твердой фазы, она остается некоторое время
постоянной или повышается перед тем, как стать постоянной (в этот момент
прекращают перемешивание), а затем снова падает. Отмечают наиболее высокую
температуру, остающуюся короткое время постоянной с начала затвердевания
вещества. Эту температуру и принимают за температуру затвердевания.
Если вещество остается жидким при ожидаемой температуре затвердевания,
его охлаждают на 1-2°С ниже ожидаемой температуры и вызывают затвердевание
внесением кристаллика испытуемого вещества. Для веществ, имеющих высокую
температуру затвердевания, определение можно проводить по методу Жукова
(ГОСТ 4255—75).
23
24.
Прибор Жукова24
25. Определение температурных пределов перегонки
Под температурными пределами перегонки подразумеваютинтервал между начальной и конечной температурой кипения при
нормальном давлении 101,3 кПа (760 мм рт. ст.).
Начальной температурой кипения считают температуру, при
которой в приемник перегнались первые 5 капель жидкости.
Конечной температурой кипения считают температуру, при которой
в приемник перешло 95% жидкости.
25
26.
Определение производят в приборе, состоящем из следующих частей.1. Колба для перегонки из термостойкого стекла вместимостью 100 мл с
отводной трубкой, отходящей от середины горла под углом к его нижней части
75±2° (ГОСТ 10394—72).
Наружный диаметр шара, мм
65±2
Внутренний диаметр горла, мм
16±1
Общая высота колбы, мм
215±3
Высота горла, мм
150±3
Длина отводной трубки, мм
100±3
2. Холодильник из термостойкого стекла с вставной трубкой (ГОСТ 9499—
70).
Общая длина вставной трубки, мм
530±15
Длина кожуха, мм
400±10
Длина воронки вставной трубки, мм
60±5
Внутренний диаметр воронки, мм
14,5±1
Наружный диаметр вставной трубки, мм
17±1
Угол среза вставной трубки
45±3˚
Конец вставной трубки, входящий в приемник, должен быть изогнут; можно
также пользоваться алонжем. При работе с жидкостями, кипящими при
температуре ниже 150°С, применяют водяное охлаждение. Для жидкостей,
кипящих при температуре выше 150°С, достаточно воздушного охлаждения. 26
27.
3.Приемник, в качестве которого может служить цилиндр вместимостью 50 мл
с ценой деления 1 мл (ГОСТ 1770—74 Е).
4.
Термометр укороченный с ценой деления шкалы 0,5°С (ГОСТ 215—73).
5.
Квадратный асбестовый картон (ГОСТ 12871—67) с длиной стороны не менее
12 см и толщиной не менее 3 мм, с круглым отверстием в центре диаметром от 2
до 3 см.
6.
Источник нагрева: газовая горелка или другой источник, обеспечивающий
необходимую температуру, безопасность и контроль за перегонкой.
7.
Два штатива: один снабжен лапкой и кольцом для укрепления колбы, другой
— лапкой для укрепления холодильника.
27
28.
Методика определения. В горло колбы вставляют термометр с помощьюхорошо подобранной корковой пробки таким образом, чтобы верхний край
ртутного шарика находился на 1 см ниже нижнего края отводного отверстия,
затем колбу укрепляют с помощью лапки на штативе так, чтобы она плотно
закрывала отверстие асбестового картона, лежащего на кольце, прикрепленном к
тому же штативу.
К отводной трубке колбы с помощью нормального шлифа или корковой
пробки присоединяют холодильник (укрепленный на штативе лапкой) так, чтобы
конец отводной трубки входил в трубку холодильника не менее чем на 3—4 см, но
не достигал суженной части. Собранный таким образом прибор в целях
безопасности устанавливают на противне с песком.
Отмеривают 50 мл исследуемой жидкости цилиндром, используемым в
качестве приемника, и переливают в колбу, пользуясь воронкой, чтобы жидкость
не попадала на стенки колбы и в особенности в отводную трубку. В колбу
опускают несколько тонких запаянных с одного конца капилляров. Приемник
помещают так, чтобы изогнутый конец холодильника входил в него на 2,5 см.
Начинают нагревание колбы и отмечают начальную температуру кипения;
затем приемник придвигают к концу холодильника так, чтобы последний касался
его стенки, и продолжают нагревание таким образом, чтобы в минуту
перегонялось от 3 до 4 мл жидкости. Перегоняют требуемый объем жидкости,
отмечая конечную температуру кипения.
28
29.
Наблюдаемые температурные пределы перегонки (Тиспр) приводят кнормальному давлению 101,3 кПа (760 мм рт.ст.) по следующей формуле:
Тиспр= Т + К(Р-Р1),
где Т — наблюдаемая температура; Р — нормальное барометрическое
давление (101,3 кПа); P1 — барометрическое давление во время опыта,
наблюдаемое по ртутному барометру или анероиду с учетом поправок, указанных
в поверочном свидетельстве и в инструкции по эксплуатации; К-инкремент
температуры кипения на миллиметр давления. Значение К зависит от
температуры кипения перегоняемой жидкости.
Примечания. 1. Если во время опыта давление измерялось ртутным барометром, то после
внесения поправок, указанных в поверочном свидетельстве и в инструкции по эксплуатации, оно
должно быть приведено к показаниям при температуре 0°С, для чего вычитают из показаний
барометра: 0,27 кПа (2 мм рт. ст.) при температуре окружающей среды от 13 до 20°С; 0,4 кПа (3
мм рт. ст.) при температуре окружающей среды от 21 до 28°С; 0,53 кПа (4 мм рт. ст.) при
температуре окружающей среды от 29 до 35°С.
2. Перегонку эфира следует проводить на предварительно нагретой водяной бане при
температуре от 54 до 58°С. Колбу помещают на асбестовом картоне таким образом, чтобы дно ее
полностью закрывало отверстие в картоне и было погружено в воду.
29
30.
Допустимое расхождение между результатами двухпараллельных определений 1°С.
Для определения температурных пределов перегонки
жидкостей возможно применение прибора для определения
температурных пределов перегонки (ТПП), изготовляемого
Клинским заводом «Лаборприбор» (рис. 3).
Для идентификации вещества можно применить
микрометод определения температуры кипения.
В тонкостенную стеклянную запаянную с одного конца
трубочку диаметром 3 мм и длиной около 8 см помещают
несколько капель исследуемой жидкости, чтобы образовался
слой от 1 до 1,5 см высоты. В трубочку вставляют открытым
концом вниз запаянный с одного конца капилляр длиной
около 10 см и диаметром около 1 мм. Трубочку прикрепляют
с помощью резинового колечка или тонкой проволоки к
укороченному термометру так, чтобы нижний конец
трубочки приходился на уровне середины ртутного шарика,
и термометр помещают в прибор для определения
температуры плавления.
30
31.
Нагревание ведут таким образом, чтобы температура поднималась на 2—3°Св минуту до того момента, когда из капилляра на смену отдельным воздушным
пузырькам начнет выделяться непрерывная цепочка пузырьков пара. Тогда
прекращают или уменьшают нагрев. Момент, когда прекратится выделение
пузырьков и жидкость начнет подниматься в капилляр, принимают за
температуру кипения.
Наблюдаемую температуру кипения приводят к показаниям при
нормальном давлении ртутного барометра, как указано выше.
Примечание. Если при определении температурных пределов перегонки
применяют неукороченный термометр, то следует вносить поправку на
выступающий столбик ртути. Для этого употребляют вспомогательный
термометр, помещаемый у выступающей части основного термометра так,
чтобы шарик вспомогательного термометра находился посередине между
верхней поверхностью пробки и концом столбика ртути. Исправленную
температуру вычисляют по формуле:
Тиспр = Т + 0,00016 (Т- t)N,
где Т - показание основного термометра; t- показание вспомогательного
термометра; 0,00016 - видимый коэффициент расширения ртути в стекле; N высота столбика ртути выступающей части основного термометра, выраженная в
градусах.
31
32. Определение плотности
При установлении плотности берут массу вещества определенного объема.Плотность устанавливают с помощью пикнометра или ареометра по методикам,
описанным в ГФ XI, вып. 1 (с. 24—26), строго соблюдая температурный режим,
так как плотность зависит от температуры. Обычно это достигают
термостатированием пикнометра при 20°С. Определенные интервалы значений
плотности подтверждают подлинность этилового спирта, глицерина, масла
вазелинового, вазелина, парафина твердого, галогенопроизводных углеводородов
(хлорэтила, фторотана, хлороформа), раствора формальдегида, эфира для наркоза,
амилнитрита и др. ГФ XI, вып. 1 (с. 26) рекомендует устанавливать содержание
спирта в препаратах спирта этилового 95, 90, 70 и 40%-ного по плотности, а в
лекарственных формах либо дистилляцией с последующим установлением
плотности, либо по температуре кипения водно-спиртовых растворов (в том
числе настоек).
Дистилляцию осуществляют кипячением определенных количеств
спиртоводных смесей (настоек) в колбах, герметически соединенных с
приемником. Последний представляет собой мерную колбу вместимостью 50 мл.
Собирают 48 мл отгона, доводят его температуру до 20°С и добавляют водой до
метки. Плотность отгона устанавливают пикнометром.
32
33.
3334. Определение спирта в фармацевтических препаратах
В круглодонную колбу вместимостью 200—250 мл отмеривают точноеколичество жидкости. При содержании спирта в жидкости до 20% для
определения берут 75 мл жидкости, если жидкость содержит от 20 до 50% — 50
мл, от 50% и выше — 25 мл; жидкость перед перегонкой разбавляют водой до 75
мл.
Для равномерного кипения в колбу с жидкостью помещают капилляры, пемзу
или кусочки прокаленного фарфора. Если жидкость при перегонке сильно
пенится, то добавляют фосфорную или серную кислоту (2—3 мл), хлорид
кальция, парафин или воск (2—3 г).
Приемник (мерную колбу вместимостью 50 мл) помещают в сосуд с холодной
водой, собирают около 48 мл отгона, доводят его температуру до 20°С и
добавляют воды до метки. Отгон должен быть прозрачным или слегка мутным.
Плотность отгона определяют пикнометром и по алкоголеметрическим
таблицам находят соответствующее содержание спирта в процентах по объему.
Содержание спирта в препарате (X) в процентах по объему вычисляют по
формуле:
34
35.
где 50 — объем отгона в миллилитрах; а — содержание спирта в процентахпо объему; б — объем исследуемого препарата, взятый для отгона, в
миллилитрах.
При содержании в жидкости эфира, эфирных масел, хлороформа, камфоры к
ней добавляют в делительной воронке равный объем насыщенного раствора
натрия хлорида и такой же объем петролейного эфира. Смесь взбалтывают в
течение 3 мин. После разделения слоев спиртоводный слой сливают в другую
делительную воронку и обрабатывают таким же образом половинным
количеством петролейного эфира. Спиртоводный слой сливают в колбу для
отгона, а соединенные эфирные жидкости взбалтывают с половинным
количеством насыщенного раствора натрия хлорида, потом присоединяют к
жидкости, находящейся в колбе для отгона.
Если жидкость содержит менее 30% спирта, то высаливание производят не
раствором, а 10 г сухого натрия хлорида.
При содержании летучих кислот их нейтрализуют раствором щелочи, при
содержании летучих оснований — фосфорной или серной кислотой.
35
36.
Жидкости,содержащие
свободный
йод,
перед
дистилляцией
обрабатывают
цинковой
пылью
или
рассчитанным количеством сухого натрия тиосульфата до
обесцвечивания. Для связывания летучих сернистых
соединений прибавляют несколько капель раствора едкого
натра.
Определение содержания спирта в настойках проводят
также по температуре кипения.
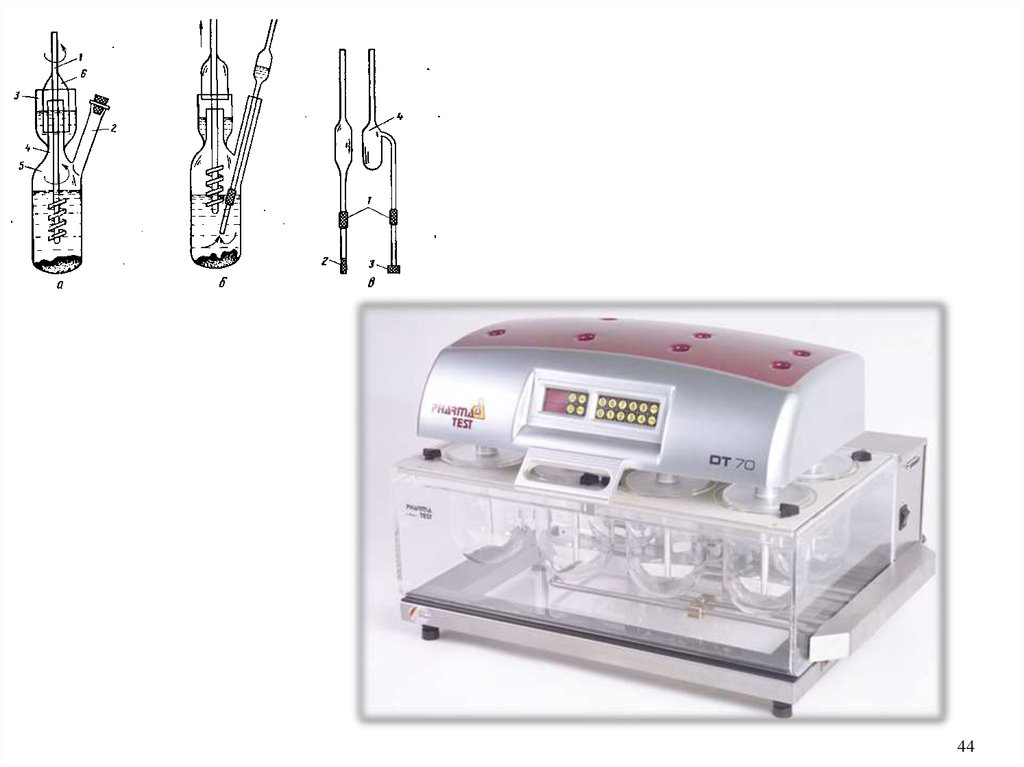
Прибор для количественного определения спирта в
настойках состоит из сосуда для кипячения 1, трубки 2 с
боковым отростком, холодильника 3 и ртутного термометра 4 с
ценой деления 0,1°С и пределом шкалы от 50 до 100°С (рис. 4).
В сосуд для кипячения наливают 40 мл настойки и для
равномерного кипения помещают капилляры, пемзу или
кусочки прокаленного фарфора. Термометр помещают в
приборе таким образом, чтобы ртутный шарик выступал над
уровнем жидкости на 2—3 мм.
36
37.
Нагревают на сетке с помощью электроплитки мощностью 200 Вт илигазовой горелки. Когда жидкость в колбе начнет закипать, с помощью реостата в 2
раза уменьшают напряжение, подаваемое на плитку. Через 5 мин после начала
кипения, когда температура становится постоянной или ее отклонение не
превышает ±0,1 °С, снимают показания термометра. Полученный результат
приводят к нормальному давлению. Если показания барометра отличаются от
1011 гПа (760 мм рт. ст.), вносят поправку на разность между наблюдаемым и
нормальным давлением 0,04°С на 1,3 гПа (1 мм рт. ст.). При давлении ниже 1011
гПа поправку прибавляют к установленной температуре, при давлении выше 1011
гПа поправку вычитают.
Пример. Температура кипения настойки пустырника 80,9°С, атмосферное
давление 1000 гПа (752 мм рт. ст.), разность давлений 1011 —1000 = 11 гПа (760
— 752 = 8 мм. рт. ст.). Поправка составляет: 0,04°С×8 = 0,32°С. К найденной
температуре кипения прибавляют поправку: (80,9 + 0,32) °С. По таблице этой
температуре кипения соответствует 66% спирта.
37
38.
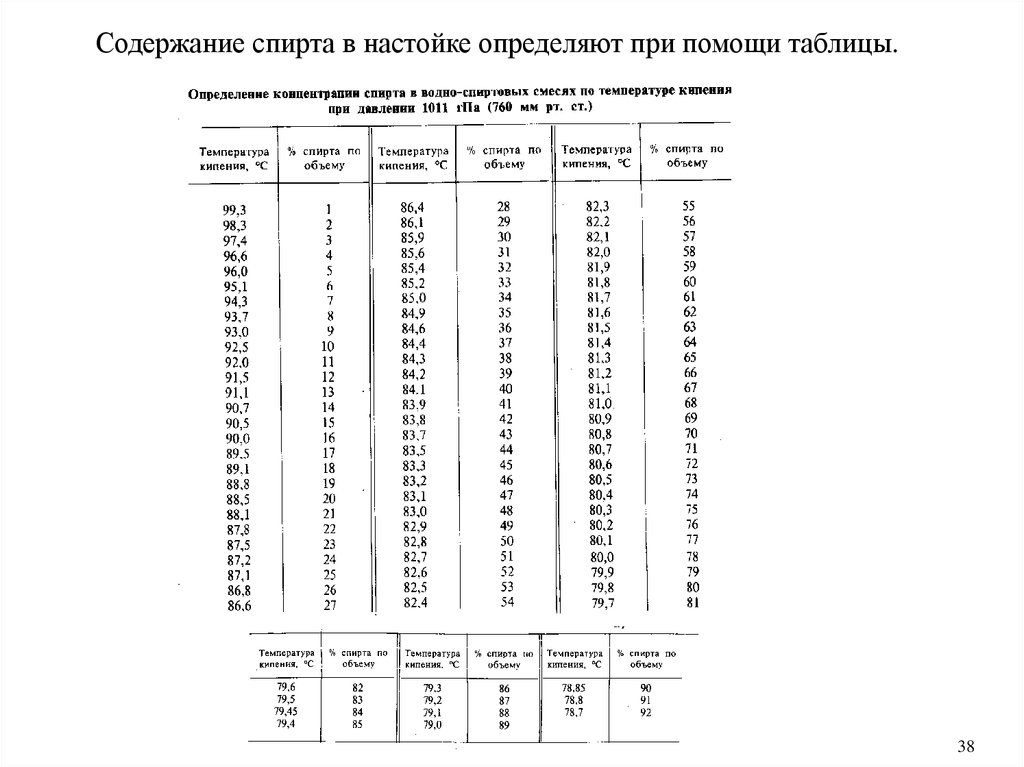
Содержание спирта в настойке определяют при помощи таблицы.38
39.
3940. Определение вязкости
Вязкость (внутреннее трение) — физическая константа, подтверждающаяподлинность жидких лекарственных веществ. Различают динамическую
(абсолютную), кинематическую, относительную, удельную, приведенную и
характеристическую вязкость. Каждая из них имеет свои единицы измерения.
Для оценки качества жидких препаратов, имеющих вязкую
консистенцию, например глицерина, вазелина, масел, обычно определяют
относительную вязкость. Она представляет собой отношение вязкости
исследуемой жидкости к вязкости воды, принятой за единицу. Для измерения
кинематической
вязкости
используют
различные
модификации
вискозиметров типа Оствальда и Уббелоде. Кинематическую вязкость обычно
выражают в м2 * с-1. Зная плотность исследуемой жидкости, можно затем
вычислить динамическую вязкость, которую выражают в Па * с.
Динамическую вязкость можно также установить с помощью ротационных
вискозиметров различных модификаций типа ''Полимер РПЭ-1И или
микрореометров серии ВИР. На измерении скорости падения шарика в
жидкости основано устройство вискозиметров типа Гепплера. Они позволяют
установить
динамическую
вязкость.
Все
приборы
должны
термостатироваться, так как вязкость в значительной степени зависит от
температуры испытуемой жидкости.
40
41.
4142. Определение растворимости
Методика определения растворимости по ГФ XI основана на том, что навескапредварительно растертого (в необходимых случаях) препарата вносится в
отмеренный объем растворителя и непрерывно перемешивается в течение 10 мин
при (20±2)°С.
В ГФ XI, вып. 1 (с. 149) включен метод фазовой растворимости, который дает
возможность
осуществлять
количественную
оценку
степени
чистоты
лекарственного вещества путем точных измерений значений растворимости. Этот
метод основан на правиле фаз Гиббса, которое устанавливает зависимость между
числом фаз и числом компонентов в условиях равновесия.
42
43.
Суть установления фазовой растворимости заключается в последовательномприбавлении увеличивающейся массы препарата к постоянному объему
растворителя.
Для
достижения
состояния
равновесия
смесь
подвергают
длительному встряхиванию при постоянной температуре, а затем с помощью
диаграмм определяют содержание растворенного лекарственного вещества, т.е.
устанавливают, является ли испытуемый препарат индивидуальным веществом
или смесью. Метод фазовой растворимости отличается объективностью, не
требует для выполнения дорогостоящего оборудования, знания природы и
структуры примесей. Это позволяет использовать его для качественного и
количественного анализов, а также для изучения стабильности и получения
очищенных образцов препаратов (до степени чистоты 99,5%), Важное
достоинство
метода
—
возможность
отличать
оптические
изомеры
и
полиморфные формы лекарственных веществ. Метод применим ко всем видам
соединений, которые образуют истинные растворы.
43