





































 medicine
medicineSimilar presentations:







")


The leukemia
1.
THE LEUKEMIABy Fineman Riva, MD
Hematology and BMT dpt, RAMBAM Med.
Center, HAIFA, ISRAEL
2.
TERMINOLOGYA malignant proliferation of monoclonal
hematopoetic cells with accumulation of
abnormal immature cells which replace a
normal bone marrow.
Those cells retain the capacity to divide
and proliferate, but lose the ability to
differentiate terminally into mature
hematopoetic cells and to die in a
programmed cell death (apoptosis)
3.
LeukemiasA very heterogeneic group of
disorders, can be classified on a basis
of clinical course as acute or chronic,
on a basis of cell lineage –
myelogenous or lymphatic
AML – Acute Myeloblastic Leukemia
ALL – Acute Lymphoblastic Leukemia
CML – Chronic Myeloid Leukemia
CLL – Chronic Lymphocytic Leukemia
4.
AML – FAB classificationM0 – undifferentiated
M1 – early myeloblastic
M2 – myelocytic
M3 – promyelocytic
M4 – myelomonocytic
M5 – monocytic/monoblastic
M6 – erythroid
M7 - megacariocytic
5.
FAB classificationThis classification is based mostly on
morphology and immunophenotyping of
the blasts
Has clinical and prognostic
correlation, but not consistent
Updates should include cytogenetic
features
6.
CytogeneticsCytogenetics is the most important
prognostic feature of AML
“Favorable” – M2 with t(8;21), M3
with t(15;17), M4eo with (inv 16)
Regular – normal caryotype
Unfavorable 11q23, 7q-, 5q-, trisomy
8, FLT-3 polymorphism, etc.
7.
AML – WHO classificationAML with recurrent cytogenetic
translocations – M2 with t(8;21), M3 with
t(15;17) and variants, M4eo with (inv16),
AML with 11q23 abnormalities
AML with multilineage dysplasia MDS
AML or MDS therapy related (alkylating
agents, epydiphylotoxin, other)
FAB subtypes without other features
Acute biphenotypic leukemia
8.
ALLThe FAB classification is not in use
Is classified by the phenotype of the
blasts – early B-lymphoblastic, Tlymphoblastic, mature Blymphoblastic (Burkitt leukemia)
“Favorable” cytogenetics – t(12;21) in
children
Unfavorable – Ph chromosome t(9;22),
11q23, t(4;11)
9.
ETIOLOGYEnvironment: irradiation, chemotherapeutic
agents, organic solvents – benzene etc.
Genetic diseases: neurofibromatosis,
Wiscott-Aldrich synd., defective DNA
repair – Fanconi, Down synd.
Acquired disorders: Aplastic Anemia, PNH
MOST OF THE CASES APPEAR WITH
NO APPARENT RISK FACTORS!!!
10.
CLINICAL FEAURESAML – 1.2% of all cancer deaths in US
(about 9200 new cases per year), the
incidence increases with age. In
adults represents 90% of acute
leukemias
ALL less prevalent in adults, the
incidence increases in seventh and
eighth decades of age
11.
CLINICAL FEATURESThe presenting signs are not specific:
Anemia – pallor, weakness, dispnoea
Neutropenia – fever, infections
Thombocytopenia – bleeding, petechiae
Extramedullary - mild splenomegaly, skin
involvement – leukemia cutis, chloromas,
gingival hyperplasia more in monocytic or
M2 with t(8;21), CNS more in ALL
12.
LABORATORYLeukocytosis with blasts
Metabolic and electrolyte
derangement hyperuricemia,
hyperkalemia, hyperphosphatemia –
tumor lysis syndrome
Coagulopathy – DIC typical to APL
13.
DIAGNOSISBlasts in blood or bone marrow smear,
Auer rods pathognomonic to AML
Immunohistochemistry – peroxidase
(AML), non specific esterase
(monocytic), PAS (lymphoid, erytroid),
acid phosphatase (lymphoid,
erythroid, megacariocytic)
14.
ImmunophenotypingCD – Cluster Designation, molecules on the
surface of the cell, characteristic to each
cell type, identified by monoclonal Ab.
CD33, CD34, CD11, CD13, CD14 – myeloid
CD41, CD61 – megakaryocytic
Glycophorin A – erythroid
CD2, CD3, CD4, CD7, CD8 – T lymphocytes
CD10, CD19, CD20, CD22 – B lymphocytes
15.
AML - TREATMENTAML – induction with ARA-C and
daunorubicin (7:3); consolidations
with HIDAC and others, autologous
HSCT, allogeneic HSCT, monoclonal
Ab – conjugated anti-CD33
(Gemtuzumab Ozogamycine)
APL – ATRA, chemotherapy, Arsenic
16.
ALL - TREATMENTProtocols based on treatment of
childhood ALL, prolonged and
intensive therapy with CNS
prophylaxis and maintenance
Autologous HSCT
Allogeneic HSCT
17.
CMLA clonal expansion of
hematopoetic progenitors,
characterized clinically by
myeloid hyperplasia, leukocytosis
with basophilia and splenomegaly
18.
CMLA phasic disease – chronic phase,
accelerated phase, blast crisis
Incidence – 1-2:100000
15-20% of leukemias in adults
Median age at diagnosis – 65 years
incidence in survivors of atomic
bomb in Hiroshima and Nagasaki,
atomic accident in Chernobyl
19.
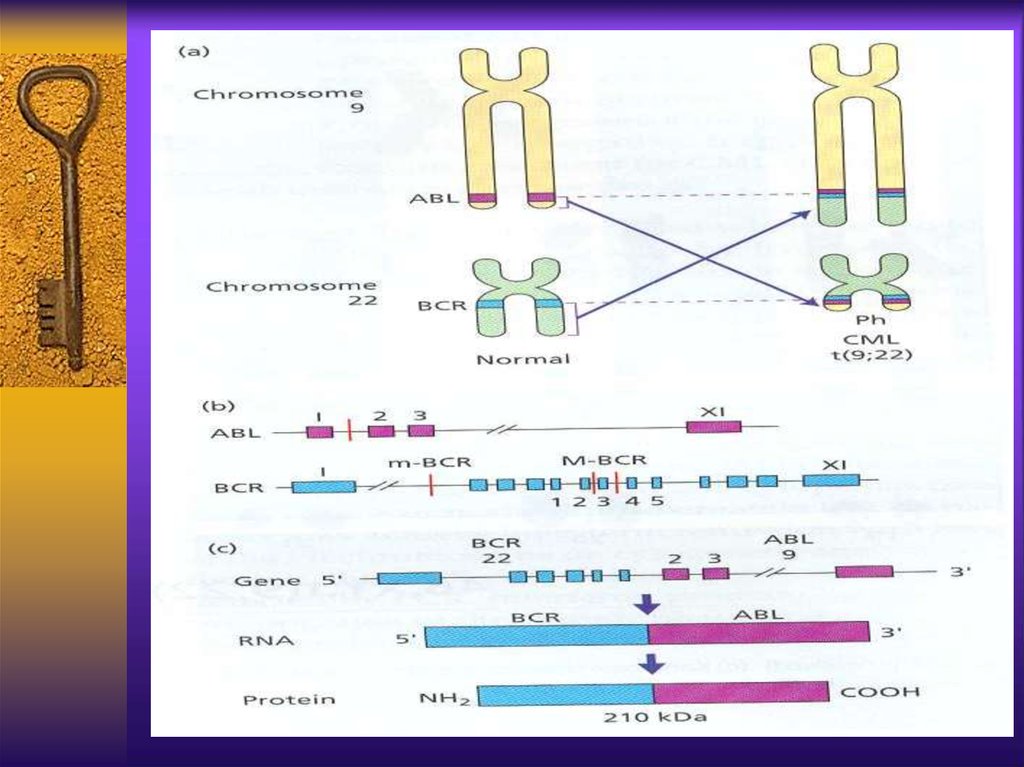
CML - cytogeneticsThe first malignancy in which the link
between a chromosomal abnormality and
leukemogenesis was established
The Philadelphia chromosome – an
abnormally short chromosome 22
The t(9;22) results from translocation of
c-abl gene from chromosome 9 to bcr gene
on chromosome 22, the new fusion gene –
bcr/abl encodes a chimeric protein with
strong unregulated tyrosine kinase activity
20.
21.
CMLPhiladelphia chromosome –
a short chromosome
22discovered at 1960 by
Novel and Henderford
First chromosomal
Abnormality connected to
malignancy
Caused by translocation
t(9;22)(q34;q11)
Results in oncogen bcr/Abl
that codes for a protein an
unregulated Tyrosine
Kinase
22.
23.
CML pathogenesisThe normal product of Abl gene is a protein
of 145kd with a week tyrosine kinase
activity, strictly regulated and important in
cell cycle regulation
The bcr/Abl product is 190,210 or 230kd
protein with strong and autonomous TK
activity, can causecell proliferation and
malignant transformation and inhibit
apoptosis. It’s substrate is oncogen ras
which inhibits tumor suppressor gene p-53
24.
Clinical featuresMost patients are asymptomatic at
diagnosis
Splenomegaly symptoms, anemia,
hepatomegaly, purpura, constitutional
symptoms – fatigue, anorexia, weight
loss,sweats,low grade fever,
hyperleukocytosis,bone pain (rare in
chronic phase), priapism
25.
LaboratoryPeripheral blood :
leukocytosis with
“left shift”,
basophillia,
eeosinophilia,
thrombocytosis,
anemia
Bone marrow:
myeloid (M:E 3:1),
blasts 10%, no
dysplasia, abundant
megacaryocytes,
fibrosis,
monocytes 3%
26.
LaboratoryLAP (leukocyte alkaline phosphatase)
Transcobalamine
Uric acid
Cytogenetics - Ph+ {t(9;22)}
Molecular - bcr/abl +
Gene expression pattern
(experimental)
27.
Accelerated PhaseLeukocytosis under treatment
Basophilia (>20% basophils and eosinophils
>10% blasts in peripheral blood
>20% blasts + promyelocytes in marrow
Thrombocytosis
Additional chromosomal abnormalities
28.
BLAST CRISISDevelopes in 75-80% of patients
Median time from diagnosis 3-5 years
constitutional symptoms, bone pain,
extramedullary (skin, lymph nodes, CNS)
>30% blasts in bone marrow
Additional chromosomal abnormalities
50% - myeloid, 25% - lymphoid, 25% biphenotypic
29.
TREATMENTTyrosine kinase inhibitors - glyvec (imatinib
mesylate), nilotinib, dasatinib etc., major
cytogenetic and molecular responses in
over 80%, changed a natural history of the
disease
Chemotherapy – hydrea, busulphan, ARA-C
-Interferon - 15% cytogenetic response
with prolonged survival
Allogeneic bone marrow transplant, 45-70%
long term survival, curative. In TKI`s
resistant or intolerant cases.
30.
CLLA progressive accumulation of
functionally incompetent mature
lymphocytes
15-20% of all leukemias, M:F=1.7:1
>95% B-CLL; 2-5% T-CLL
In people > 70, incidence 20/100000,
median age at diagnosis 55
31.
CLLFrequent family history of CLL, other
B-cell malignancies, autoimmune
disorders
No other risk factors
The cells overexpress bcl-2, an
antiapoptotic gene, lack of apoptosis
32.
CLLImmunophenotyping: B-cell markers CD19,
CD20, CD21, CD23, ; T-cell marker CD5 is
a characteristic finding.
ZAP-70, IgVH mutational status
Chromosomal abnormalities found by FISH
in 60%, most frequent chr. 13, 12, 11,
17(p53)
Chr. 13 – good prognosis, chr. 11, 12, 17 –
bad prognosis, chemotherapy resistance,
short remissions, short survival
33.
Clinical ManifestationsAutoimmune features - Coomb’s+
hemolytic anemia, ITP
Recurrent infections - due to
hypogammaglobulinemia
Symptoms: weakness, weight loss,
night sweats, fever
Physical examination:
lymphadenopathy, hepatomegaly,
splenomegaly
34.
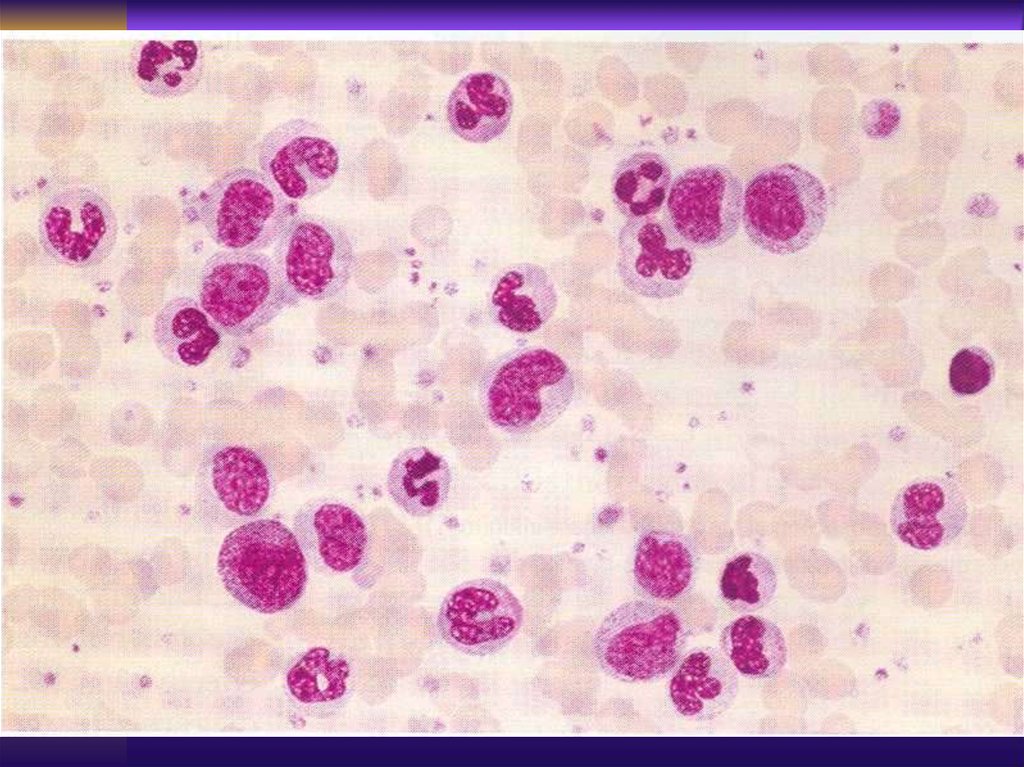
Laboratory Findings>5000 mature appearing lymphocytes
Anemia, thrombocytopenia
Bone marrow - infiltration by same
lymphocytes, decrease in myeloid and
erythroid precursors, (if ITP or
AIHA - abundant megakariocytes and
erythroid lineage
“Smudged cells” - Gumprecht cells
35.
Diagnostic CriteriaAbsolute lymphocytosis >5000/ml on
few consecutive tests
At least 30% lymphocytes in normoor hypercellular marrow
Monoclonal B-cell phenotype, CD5+
36.
CLL - Staging - Rai SystemStage 0 - lymphocytosis blood,marrow
Stage 1 - lymphocytosis + lymph nodes
Stage 2 - St.0-1 + enlarged liver or
spleen
Stage 3 - all above + anemia
Stage 4 - all above + thrombopenia
37.
CLL - Staging BinetStage A - lymphocytosis and two or
less areas of enlarged lymph nodes
Stage B - as A with three or more
areas of lymph node enlargement
Stage C - as A or B with anemia or
thrombocytopenia
38.
CLL -TreatmentRai st. 0-2 or Binet st. A-B observe
every 3-6 months, treat if disease
progress, short doubling time,
symptomatic, recurrent infections,
ITP, AIHA
Advanced stage, symptomatic needs
treatment at diagnosis
39.
Treatment OptionsChemotherapy - steroids, alkylating agents
steroids, purine analogues - fludarabine,
combinations
Monoclonal antibodies - anti CD20
(Rituximab), anti CD52 (Campath 1H) ±
chemotherapy, anti CD23
High dose therapy with hematopoetic stem
cell transplantation - autologous, allogeneic,
low intensity (“mini-transplant”)
40.
CLL - PrognosisExtremely variable - some have progressive
course and die within 2-3 years, some have
indolent disease with 10-20 years survival
The prognostic factors are - stage at
diagnosis, cytogenetics, molecular –
mutation status, ZAP-70, CD38 expression;
morphology – diffuse bone marrow
infiltration, large cells (prolymphocytes),
T-lineage, B-symptoms, high LDH, short
doubling time
41.
Richter’s SyndromeIn 3-5% the disease undergoes a
transformation into aggressive
lymphoma - diffuse large cell or
immunoblastic
Severe B-symptoms, increased LDH,
lymphadenopathy
The prognosis is poore, median
survival <6 months
42.
Second MalignanciesIncidence of 8.9% (28% increased
risk) of second malignancy
Most frequent cancers associated
with CLL are - skin,lung,
gastrointestinal tumors (ca of colon)
There is no relationship between the
course of CLL, it’s treatment and the
incidence of second cancers