






































 software
softwareSimilar presentations:










Next-generation sequencing RNA-Seq. Анализ
1.
Next-generation sequencingRNA-Seq
RNA-Seq “Анализ”
Первичный анализ и как его делать (ошибки, тримминг и
фильтры).
Что такое вторичный анализ и для чего он нужен?
(картирование, форматы файлов, контроль). Анализ
транскриптома. Анализ микроРНК.
«Агентство Химэксперт»
2.
Шаг 5: Анализ данных3.
Особенности программного обеспечениядля анализа данных NGS
Модульная организация (за каждую часть анализа отвечает своя небольшая
«подпрограмма»)
Для получения результата подпрограммы должны выстраиваться в цепочки,
которые называются pipeline. Результат одной подпрограммы в цепочке
является исходными данными для следующей подпрограммы.
Каждый компонент pipeline имеет множество настроек. «Правильные»
настройки зависят от результата, который требуется получить, и часто
неизвестны заранее.
Большинство подпрограмм созданы для операционной системы Linux и не
имеют интерфейса пользователя.
Анализ данных NGS. Брагин Антон, Sequoia Genetics. 2013.
4.
Анализ данныхA
B
C
ДНК
Приготовление
библиотеки
1
Приготовление
матрицы
2
D
E
Данные переносятся с PGM
на сервер
Перевод сигнала в сиквенс
Секвенирование
3
Анализ
данных
Оценка качества сиквенса и
анализ данных в программе
Torrent suite
5.
Анализ данных Ion TorrentОпределение положений на чипе, в которых находятся
последовательности
Перевод последовательности сигналов в последовательность
нуклеотидов
Фильтрация по качеству
• Выравнивание на референсный геном
• Поиск генетических вариантов
Анализ данных NGS. Брагин Антон, Sequoia Genetics. 2013.
6.
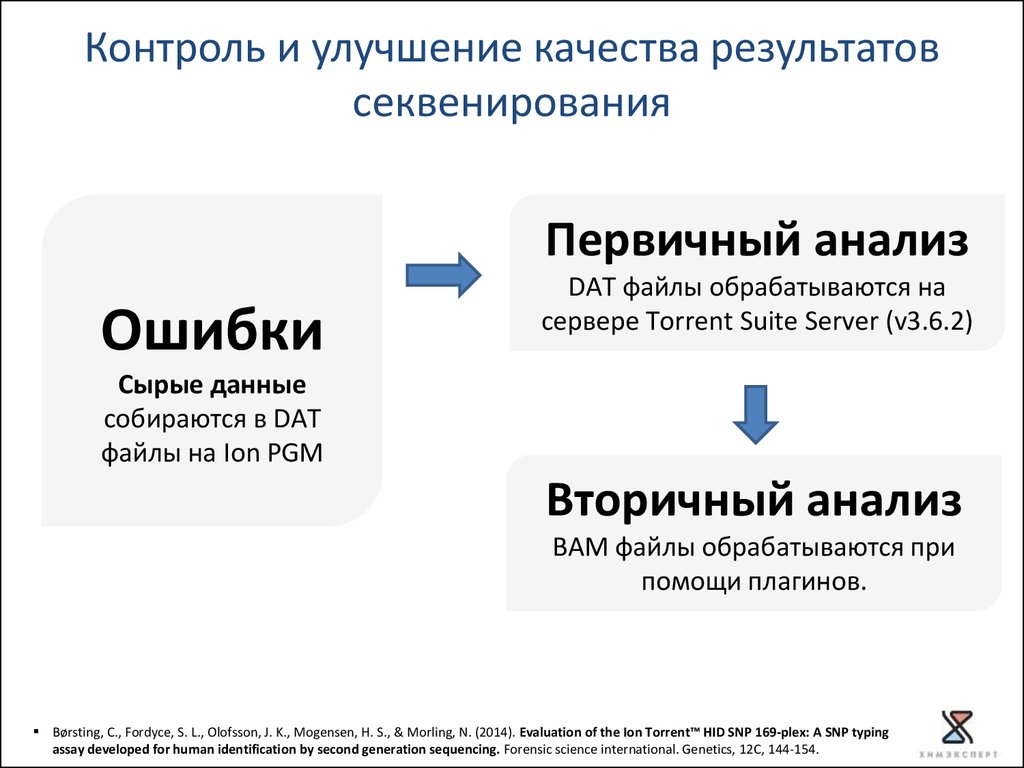
Контроль и улучшение качества результатовсеквенирования
Ошибки случаются…
Эксперимент. Ошибки постановки эксперимента
Биоинформатика. Ошибки в чтениях возникают из-за неточности работы
секвенатора
Barry Merriman, Ion Torrent R&D Team, Jonathan M. Rothberg. Progress in Ion Torrent semiconductor chip based sequencing
Electrophoresis 2012, 33, 3397–3417 3397
7.
Контроль и улучшение качества результатовсеквенирования
Первичный анализ
Ошибки
DAT файлы обрабатываются на
сервере Torrent Suite Server (v3.6.2)
Сырые данные
собираются в DAT
файлы на Ion PGM
Вторичный анализ
BAM файлы обрабатываются при
помощи плагинов.
Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H. S., & Morling, N. (2014). Evaluation of the Ion Torrent™ HID SNP 169-plex: A SNP typing
assay developed for human identification by second generation sequencing. Forensic science international. Genetics, 12C, 144-154.
8.
Возможные ошибки1) Димеры адаптеров. Адаптеры соединяются друг с другом, без
фрагмента ДНК образца
Норма
Димер адаптеров
2) Чтение сквозь. Фрагмент ДНК образца короче, чем длина чтения –
чтение захватывает часть адаптера
Слишком короткий
фрагмент
Barry Merriman, Ion Torrent R&D Team, Jonathan M. Rothberg. Progress in Ion Torrent semiconductor chip based sequencing
Electrophoresis 2012, 33, 3397–3417 3397
9.
Возможные ошибки3) Фазировка. Отдельные олигонуклеотиды в кластере со временем
начинают обгонять или отставать от остальных – секвенатору сложнее
определить букву
Чем дольше идёт прогон, тем больше будет накапливаться
отстающих и опережающих олигонуклеотидов
Barry Merriman, Ion Torrent R&D Team, Jonathan M. Rothberg. Progress in Ion Torrent semiconductor chip based sequencing
Electrophoresis 2012, 33, 3397–3417 3397
10.
Тримминг - удаление ошибок секвенирования(англ. trim – приводить в порядок)
Две задачи тримминга:
1. Удаление последовательности адаптера в чтениях
2. Отсечение с конца чтений нуклеотидов с низким качеством
(например, Q<15)
Technical Note. Trimming and Filtering
http://mendel.iontorrent.com/ion-docs/Technical-Note---Filtering-and-Trimming_6455370.html
11.
FASTQ – стандартный форматзаписи чтений
P – вероятность ошибки
Q – параметр качества (Phred Quality Score)
Phred Quality Score
Q = -10log10P
Probability of incorrect
base call
Base call accuracy
10
1 in 10
90%
20
1 in 100
99%
30
1 in 1000
99.9%
40
1 in 10,000
99.99%
50
1 in 100,000
99.999%
60
1 in 1,000,000
99.9999%
Типичные значения Q от 1 до 40
Q>20 – «хорошее качество»
1) Barry Merriman, Ion Torrent R&D Team, Jonathan M. Rothberg. Progress in Ion Torrent semiconductor chip based sequencing
Electrophoresis 2012, 33, 3397–3417 3397. 2) http://en.wikipedia.org/wiki/Phred_quality_score
12.
• Синяя линия –среднее качество
• Красная линия –
медиана
• Жёлтая рамка –
интерквартиль
(50% чтений
попадает в эти
границы)
• Чёрные засечки
– 80% чтений
попадает в эти
границы
13.
FASTQ – стандартный форматзаписи чтений
FASTQ – общепринятый формат записи чтений.
Последовательность
Качество
Barry Merriman, Ion Torrent R&D Team, Jonathan M. Rothberg. Progress in Ion Torrent semiconductor chip based sequencing
Electrophoresis 2012, 33, 3397–3417 3397
14.
FASTQ – стандартный форматзаписи чтений
Качество в формате FASTQ закодировано в ASCII – символах
Существовало несколько стандартов записи качества
Многим программам важно, чтения во Phred+33 или Phred+64
http://en.wikipedia.org/wiki/FASTQ_format
15.
Первичный анализ1. Оценивается фоновый сигнал пустых лунок и
вычитается из необработанного сигнала лунок с
ионосферами.
2. Необработанные сигналы нормализуются в
соответствии с ключевой последовательностью
(TCAG) в адаптере.
3. Проводится присваивание названий
нуклеотидам (base calling) и проводится оценка
качества каждой буквы (оценка Phred).
Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H. S., & Morling, N. (2014). Evaluation of the Ion Torrent™ HID SNP 169-plex: A SNP typing
assay developed for human identification by second generation sequencing. Forensic science international. Genetics, 12C, 144-154.
16.
Первичный анализ4. Удаляются последовательности низкого качества, димеры праймеров и
последовательности от поликлональных ионосфер.
5. Последовательности обрезаются в соответствии с баллами Phred (среднее
значение Phred на протяжении 30 п.о. <15), в соответствии с дисбалансом
сигнала (> 3% нуклеотидов в последовательности были 0.5-0.59 или 1.4-1.49
п.о. для одной буквы и 1.5-1.59 или 2.4-2.49 п.о. для двух букв) и по
последовательности нуклеотидов в 3‘ адаптере.
6. Последовательности выравниваются на референс (эталонная
последовательность генома человека – 19) и генерируется бинарный файл с
координатами выравнивания (BAM файл)
Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H. S., & Morling, N. (2014). Evaluation of the Ion Torrent™ HID SNP 169-plex: A SNP typing
assay developed for human identification by second generation sequencing. Forensic science international. Genetics, 12C, 144-154.
17.
Фильтры для ридовВарианты фильтрации прочтений:
1. Удаление коротких прочтений
2. Удаление димеров адаптеров
3. Удаление прочтений без ключевой последовательности (TCAG в
обычных ридах, ATCG в контроле)
4. Удаление прочтений с зашкаливающим (off-scale) сигналом
5. Удаление поликлональных прочтений
Technical Note. Trimming and Filtering
http://mendel.iontorrent.com/ion-docs/Technical-Note---Filtering-and-Trimming_6455370.html
18.
Фильтр на удаление поликлональных прочтенийTechnical Note. Trimming and Filtering
http://mendel.iontorrent.com/ion-docs/Technical-Note---Filtering-and-Trimming_6455370.html
19.
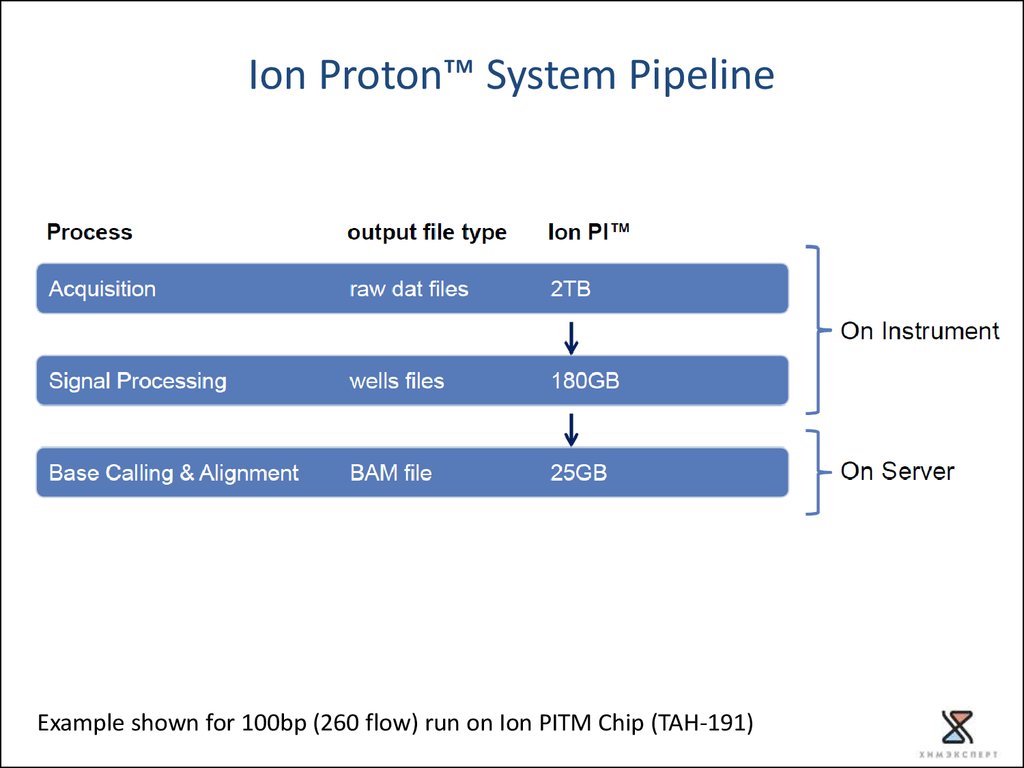
Torrent Suite™ Data Analysis Flow20.
Ion Proton™ System PipelineExample shown for 100bp (260 flow) run on Ion PITM Chip (TAH-191)
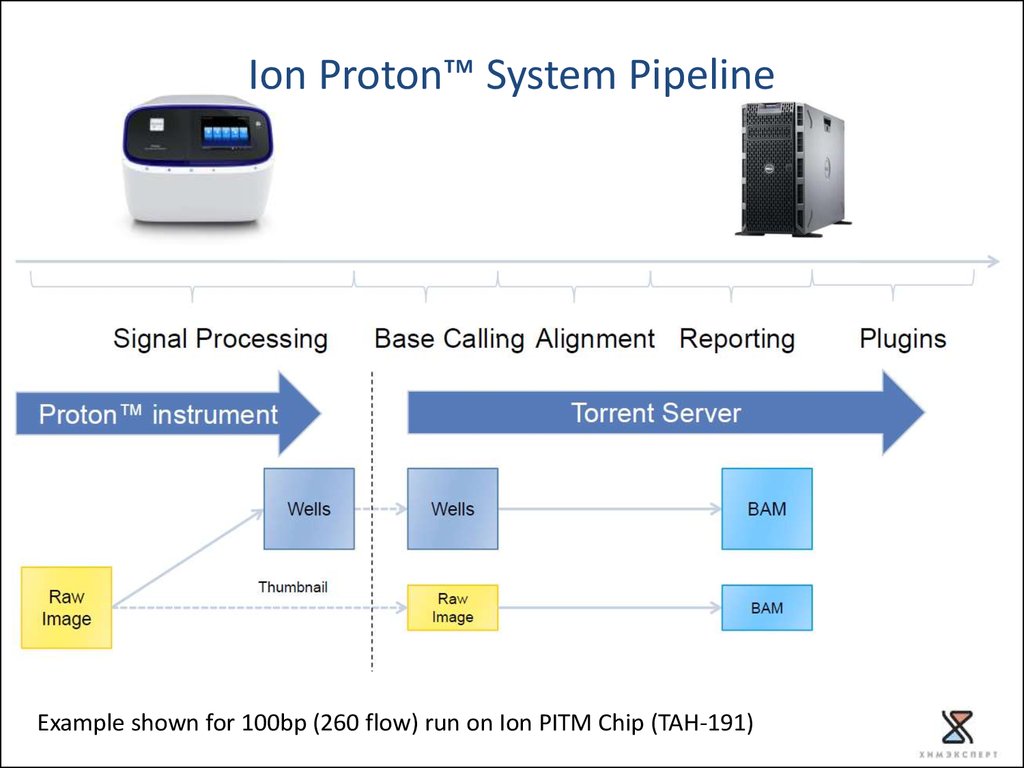
21.
Ion Proton™ System PipelineExample shown for 100bp (260 flow) run on Ion PITM Chip (TAH-191)
22.
Вторичный анализ(Обработка BAM файлов при помощи плагинов)
Methods, tools, and pipelines
for analysis of Ion PGM™
Sequencer miRNA and gene
expression data
23.
Основные задачипри работе с прочтениями на Ion PGM™ Torrent Server
Шаг 1: Тримминг по качеству 3’ конца и Ion P1B адаптеру
Шаг 2: Оценка и контроль качества
Шаг 3: Картирование на референсный геном или транскриптом
Шаг 4: Подсчет картированных прочтений
Шаг 5: Статистический анализ
24.
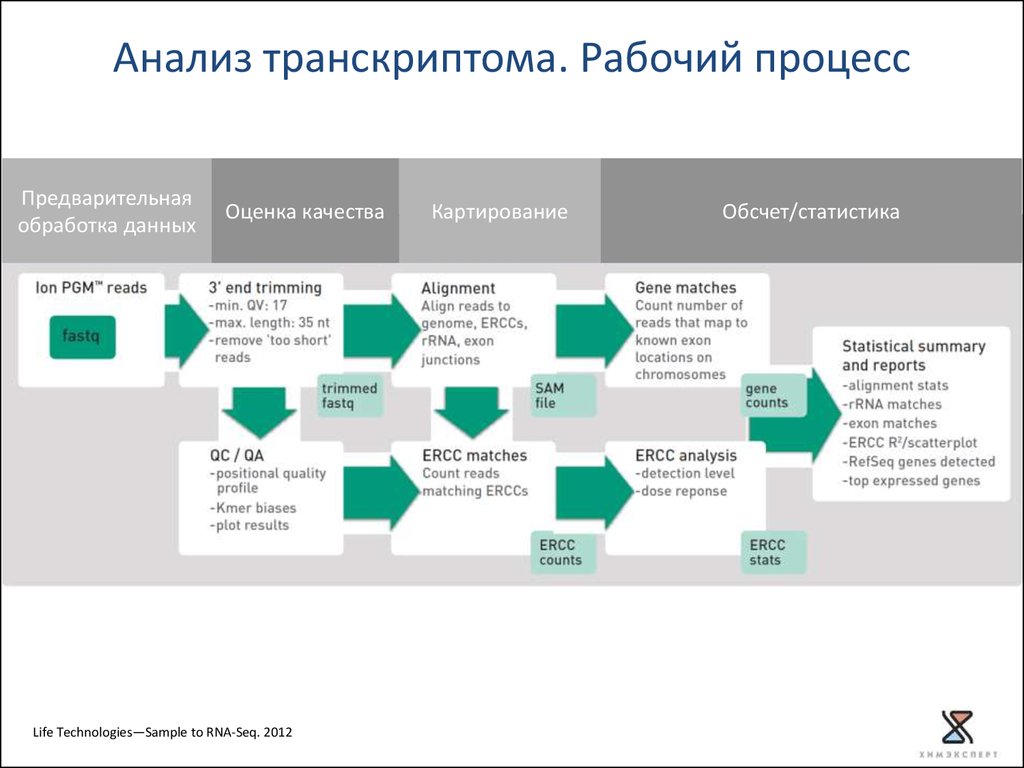
Анализ транскриптома. Рабочий процессПредварительная
обработка данных
Оценка качества
Life Technologies—Sample to RNA-Seq. 2012
Картирование
Обсчет/статистика
25. Ion Proton™ System Enables High Quality Transcriptome Analysis with >80M Reads per Run
Ion Proton™ System Enables High Quality Transcriptome Analysis with>80M Reads per Run
Ion Proton™ Runs with MAQC Universal Human Reference (UHR)
120 000 000
100 000 000
Total Reads
Mapped Reads
65% Avg
Mapping
Rate
80 000 000
60 000 000
40 000 000
20 000 000
0
Run 1
Run 2
Run 3
Run 4
Run 5
Run 6
# of refGenes Detected
16,257
16,767
16,525
16,560
16,829
16,629
% of refGenes Detected
75.4%
77.8%
76.6%
76.8%
78.1%
77.1%
26.
Шаг 1: Тримминг по качеству3’ конца и Ion P1B адаптеру
Дополнительный шаг по обрезанию концов.
Низкое качество и фрагменты адаптерной
последовательности приводят к ошибкам картирования
или такое прочтение вовсе не картируется.
FASTX-toolkit – программный пакет набора инструментов для
обработки и оценки FASTQ файлов.
Инструмент fastq_quality_trimmer применяют таким образом,
что последовательности ниже минимального значения
качества Phred (QV) 17 при сканировании от 5 'к 3' концу
прочтения отделяются. Если длина прочтения после обрезка
падает ниже 35 оснований, то оно исключается из
дальнейшего анализа, чтобы обеспечить более высокую
специфичность при выравнивании на референсный геном.
27.
Шаг 2: Оценка и контроль качестваПрограмма FastQC использует FASTQ файл в качестве
входных данных
Per Base Sequence Quality
Per Sequence Quality Scores
Per Base Sequence Content
Per Base GC Content
Per Sequence GC Content
Per Base N Content
Sequence Length Distribution
Duplicate Sequences
Overrepresented Sequences
Overrepresented Kmers
Качество прочтений по каждому нуклеотиду
Оценка качества сиквенса
Содержание GC
Содержание недостоверных (N) оснований
Распределение длин прочтений
Повторяющиеся последовательности
Анализ представленности последовательностей
Анализ представленности Kmer-ов
Этот анализ может оказаться полезным, когда на
референсный геном картируется меньше прочтений,
чем ожидалось.
28.
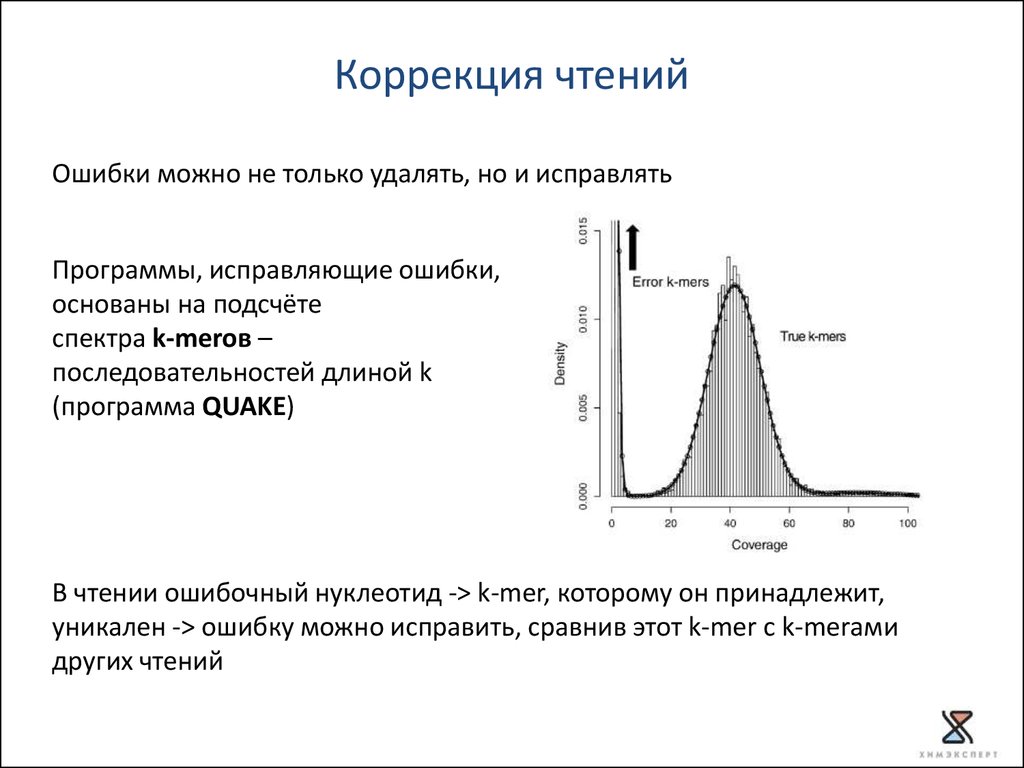
Коррекция чтенийОшибки можно не только удалять, но и исправлять
Программы, исправляющие ошибки,
основаны на подсчёте
спектра k-merов –
последовательностей длиной k
(программа QUAKE)
В чтении ошибочный нуклеотид -> k-mer, которому он принадлежит,
уникален -> ошибку можно исправить, сравнив этот k-mer с k-merами
других чтений
29.
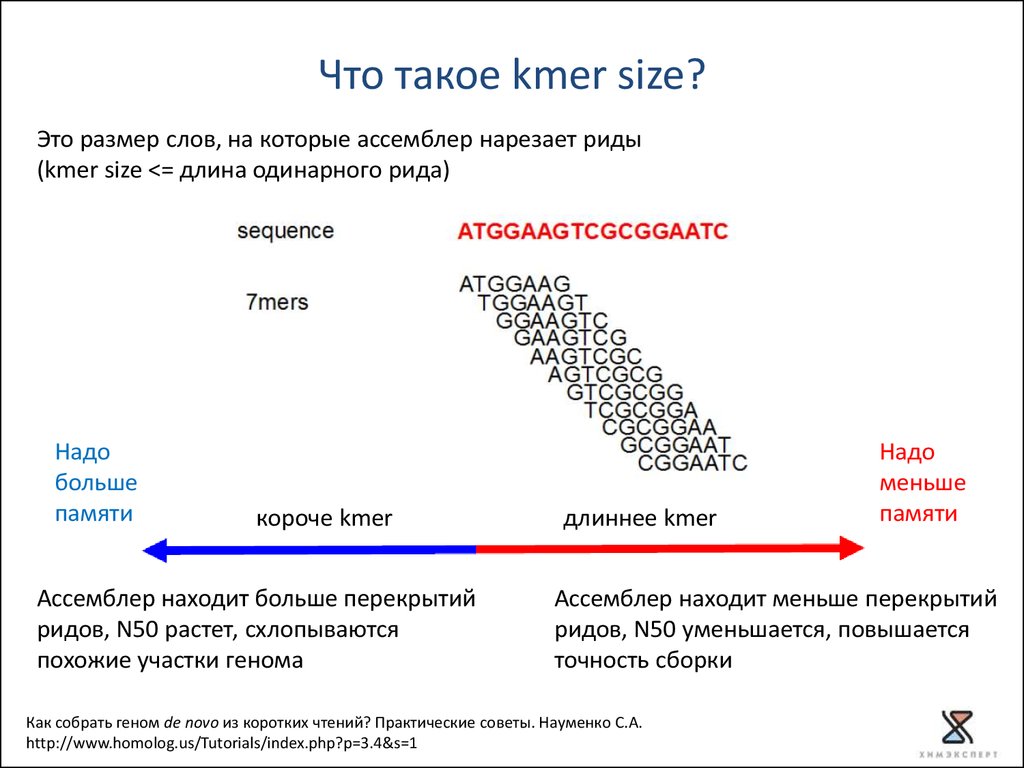
Что такое kmer size?Это размер слов, на которые ассемблер нарезает риды
(kmer size <= длина одинарного рида)
Надо
больше
памяти
короче kmer
Ассемблер находит больше перекрытий
ридов, N50 растет, схлопываются
похожие участки генома
длиннее kmer
Надо
меньше
памяти
Ассемблер находит меньше перекрытий
ридов, N50 уменьшается, повышается
точность сборки
Как собрать геном de novo из коротких чтений? Практические советы. Науменко С.А.
http://www.homolog.us/Tutorials/index.php?p=3.4&s=1
30.
Шаг 3: КартированиеПрограмма-картировщик TMAP
(The Torrent Mapping Alignment Program for Ion Torrent Data)
Для анализа транскриптома следует использовать параметр с
длиной фрагмента для анализа 18 нуклеотидов (и количество
допустимых несовпадений по умолчанию)
На выходе: файл формата SAM (Sequence Alignment Map)
Бинарный вариант этого формата: файл BAM
(файл формата BAM можно получить при помощи SamTools)
31.
SAM-формат(Sequence Alignment/Map format)
– текстовый формат, предназначенный для представления информация о
картировании чтений. Значения отдельных колонок разделяются табуляцией.
r001/1 и r001/2 спаренные риды
r003 химерный рид
r004 разделенное выравнивание
ID чтения
хромосома и
координата, куда
"легло" чтение
качество картирования
согласно картировщику
32.
BAM-формат(Binary Sequence Alignment/Map)
- Сжатый бинарный вариант формата SAM. Для быстрого
доступа к данным по выравниванию ридов.
33.
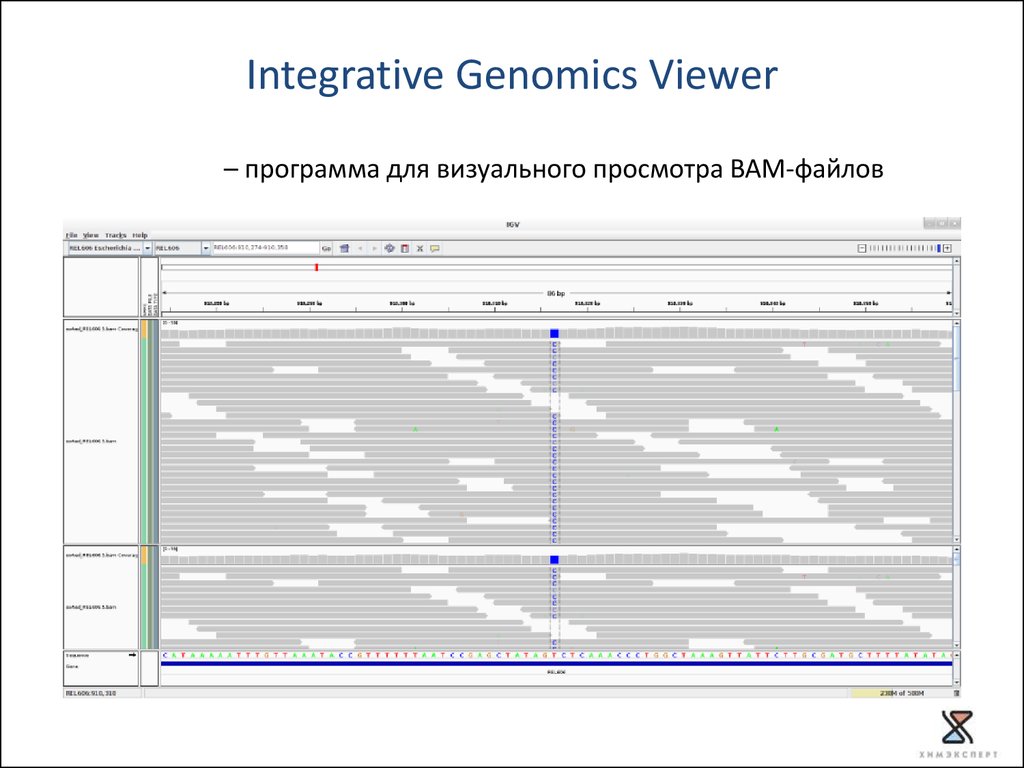
Integrative Genomics Viewer– программа для визуального просмотра BAM-файлов
34.
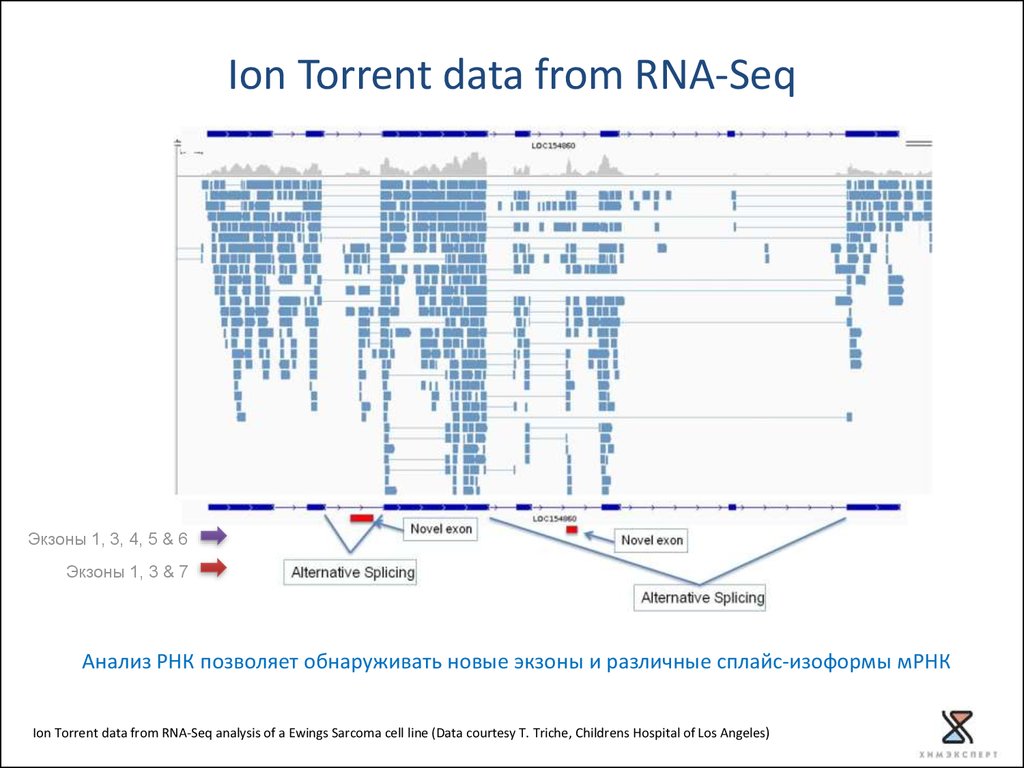
Ion Torrent data from RNA-SeqЭкзоны 1, 3, 4, 5 & 6
Экзоны 1, 3 & 7
Анализ РНК позволяет обнаруживать новые экзоны и различные сплайс-изоформы мРНК
Ion Torrent data from RNA-Seq analysis of a Ewings Sarcoma cell line (Data courtesy T. Triche, Childrens Hospital of Los Angeles)
35.
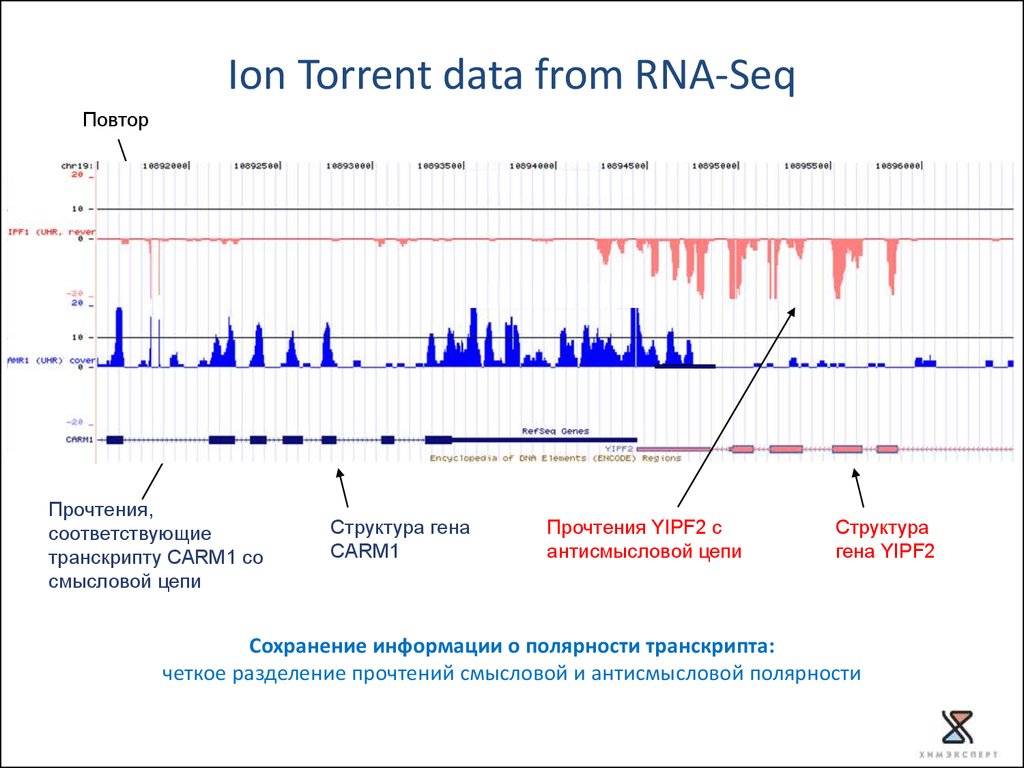
Ion Torrent data from RNA-SeqПовтор
Прочтения,
соответствующие
транскрипту CARM1 со
смысловой цепи
Структура гена
CARM1
Прочтения YIPF2 с
антисмысловой цепи
Структура
гена YIPF2
Сохранение информации о полярности транскрипта:
четкое разделение прочтений смысловой и антисмысловой полярности
36.
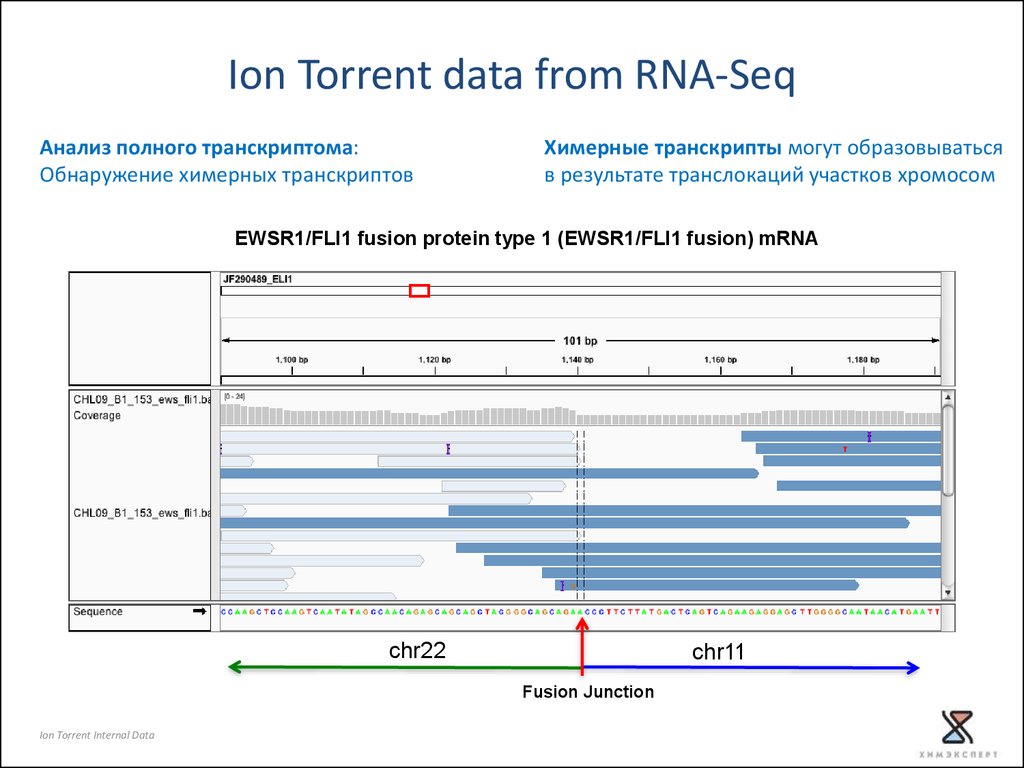
Ion Torrent data from RNA-SeqАнализ полного транскриптома:
Обнаружение химерных транскриптов
Химерные транскрипты могут образовываться
в результате транслокаций участков хромосом
EWSR1/FLI1 fusion protein type 1 (EWSR1/FLI1 fusion) mRNA
chr22
chr11
Fusion Junction
Ion Torrent Internal Data
37.
Шаг 4: Подсчет картированныхпрочтений
Скрипт htseq-count.py для подсчета картированных прочтений
Скрипт – простая программа, предназначенная для решения
конкретной проблемы «здесь и сейчас» (htseq-count.py входит
в пакет программ HTSeq Python package)
Что делает:
из SAM-файла извлекает координаты картированного
прочтения
определяет перекрывание с известными экзонами из
RefSeq.
Информацию об известных экзонах можно загрузить с сайта UCSC Genome Browser в формате файла
RefGene GTF.
Для подсчета Контроля (92 варианта последовательностей ERCC) – добавляем их в файл RefGene GTF.
38.
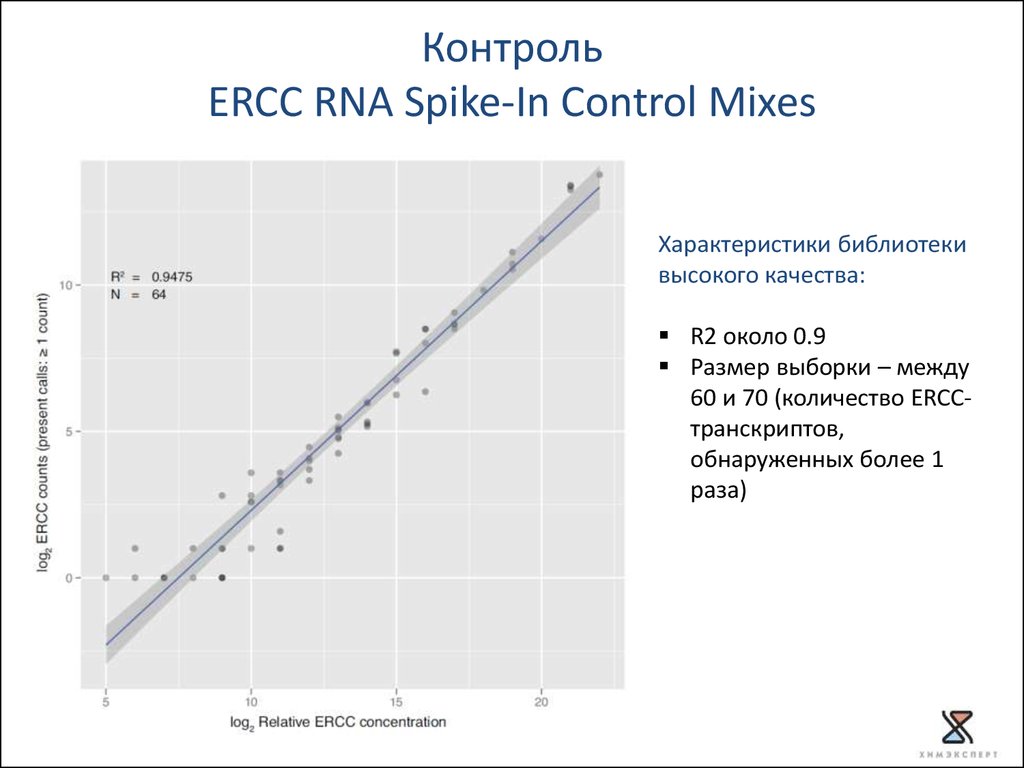
КонтрольERCC RNA Spike-In Control Mixes
92 варианта полиаденилированных транскриптов
размер транскриптов 250-2000 нукл.
«мимикрирует» под естественную эукариотическую мРНК
Spike-in РНК:
известна последовательность
известна конечная концентрация
используется для оценки точности измерений дифференциальной
экспрессии генов
39.
КонтрольERCC RNA Spike-In Control Mixes
Характеристики библиотеки
высокого качества:
R2 около 0.9
Размер выборки – между
60 и 70 (количество ERCCтранскриптов,
обнаруженных более 1
раза)
40.
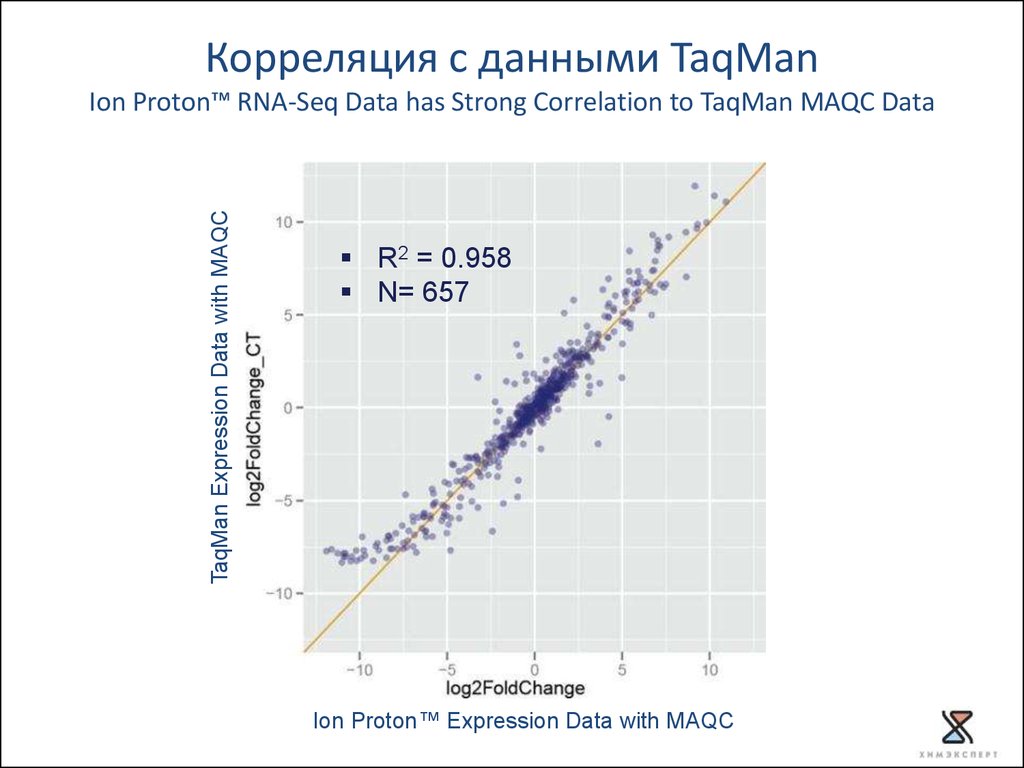
Корреляция с данными TaqManTaqMan Expression Data with MAQC
Ion Proton™ RNA-Seq Data has Strong Correlation to TaqMan MAQC Data
R2 = 0.958
N= 657
Ion Proton™ Expression Data with MAQC
41.
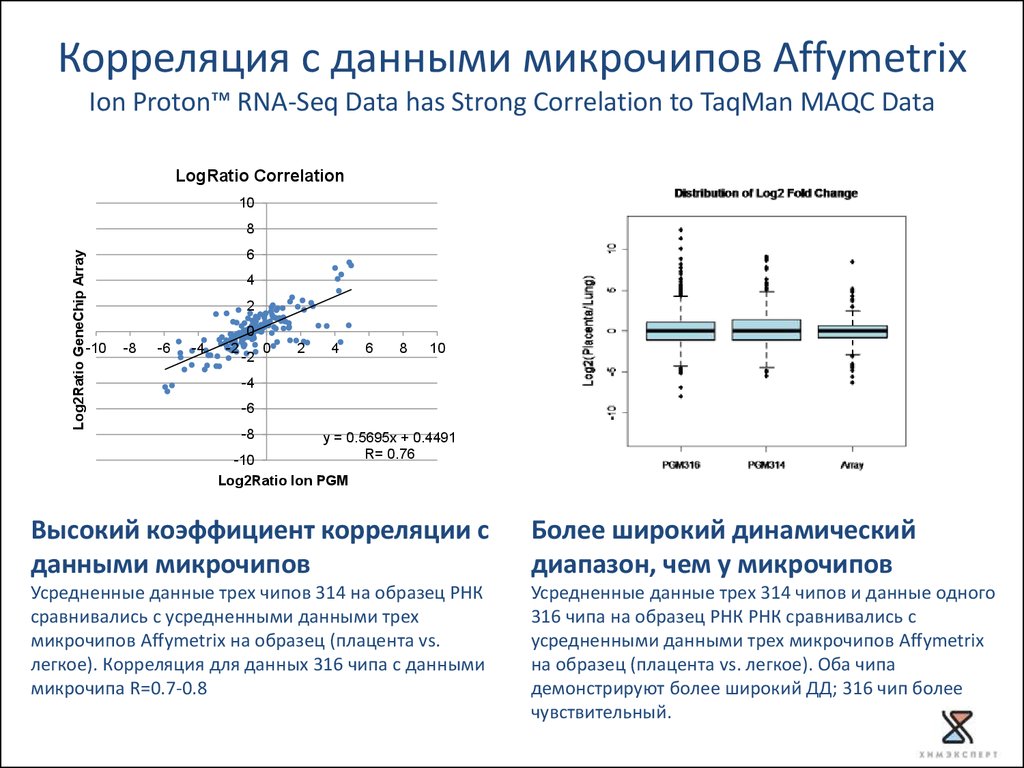
Корреляция с данными микрочипов AffymetrixIon Proton™ RNA-Seq Data has Strong Correlation to TaqMan MAQC Data
LogRatio Correlation
10
Log2Ratio GeneChip Array
8
-10
6
4
2
0
-8
-6
-4
-2
-2
0
2
4
6
8
10
-4
-6
-8
y = 0.5695x + 0.4491
R= 0.76
-10
Log2Ratio Ion PGM
Высокий коэффициент корреляции с
данными микрочипов
Более широкий динамический
диапазон, чем у микрочипов
Усредненные данные трех чипов 314 на образец РНК
сравнивались с усредненными данными трех
микрочипов Affymetrix на образец (плацента vs.
легкое). Корреляция для данных 316 чипа с данными
микрочипа R=0.7-0.8
Усредненные данные трех 314 чипов и данные одного
316 чипа на образец РНК РНК сравнивались с
усредненными данными трех микрочипов Affymetrix
на образец (плацента vs. легкое). Оба чипа
демонстрируют более широкий ДД; 316 чип более
чувствительный.
42.
Шаг 5: СтатистикаСкрипт на Perl анализирует SAM файл на базовую статистику по
картированию:
общее количество картированных ридов,
риды, картировавшиеся на экзоны,
слияния экзонов,
рРНК,
риды, картировавшиеся на неаннотированные участки генома.
Также можно провести нормализацию библиотеки.
43.
Нормализация данных RNA-SeqНормализация данных RNA-Seq необходима из-за различий в
глубине секвенирования,
длине генов,
отличий между образцами по количеству молекул,
покрытии разных мРНК (сильно различается)
Цифровая нормализация (digital normalization) – уменьшение покрытия
слишком представленных транскриптов
Нормализация с помощью программы khmer позволяет уменьшить количество чтений ~ в сто раз без
потери качества сборки.
Скорость сборки также увеличивается ~ в сто раз
Также нормализация поможет при single-cell секвенировании
44.
Статистические подходы к анализуэкспрессионных данных
Обобщенные статистические подходы к анализу транскриптомных данных и
рекомендации по данному вопросу описаны в Current Protocols in Molecular
Biology.
В случае данных абсолютной экспрессии RNA-Seq возможны прямые
сравнения величин.
Удобство прямых сравнений заключается в возможности анализа различных
комбинаций экспрессионных данных.
В случае RNA-Seq используются обобщенные линейные модели (ОЛМ), среди
которых наиболее распространены логистическая и Пуассоновская регрессия.
С развитием и внедрением RNA-Seq совершенствуются и математические
подходы, хотя они являются производными от классических ОЛМ.
45.
Анализ микроРНК. Рабочий процессПредварительная
обработка данных
Оценка качества
Картирование
Рабочий процесс анализа малых РНК для количественной оценки транскриптов микроРНК
Life Technologies—Sample to RNA-Seq. 2012
Обсчет/статистика
46.
Шаг 1: Тримминг по качеству3’ конца и Ion P1B адаптеру
FASTX-toolkit – программный пакет набора
инструментов для обработки и оценки FASTQ файлов.
fastq_quality_trimmer применяется таким же образом,
как при WT:
последовательности ниже Phred (QV) 17 при
сканировании от 5 'к 3' концу прочтения отделяются.
Но! Минимальная длина прочтения 17 оснований.
47.
Шаг 3: КартированиеSHRiMP – картировщик коротких прочтений на референс
малых РНК, содержащий последовательности шпилечных
структур микроРНК, тРНК, рРНК и 3’ адаптера.
Первый шаг картирования – шпильки микроРНК.
Предшественники микроРНК (60-90 нуклеотидов) хранятся в базе RFAM
miRBase database в FASTA формате.
Второй шаг – картирование тРНК и рРНК.
Третий шаг – картирование пропущенных ранее последовательностей
адаптера
На выходе: файл SAM формата либо файл BLAST alignment format.
48.
Шаг 4: Подсчет картированныхпрочтений
Скрипт htseq-count.py для подсчета картированных
прочтений
Что делает:
из SAM-файла извлекает координаты картированного
прочтения
Что на выходе:
Файл с колонками IDs и подсчетами для каждого
предшественника микроРНК, тРНК и рРНК
49.
Шаг 5: СтатистикаСкрипт на Perl анализирует SAM файл на базовую статистику по
картированию:
общее количество картированных ридов,
риды, картировавшиеся на экзоны,
слияния экзонов,
рРНК,
риды, картировавшиеся на неаннотированные участки генома.
Дополнительно:
Количество адаптеров без вставки
Количество обнаруженных микроРНК из базы miRBase
50.
Анализ данных на сервере Torrent ServerTorrent Server
Torrent Browser
Оценка качества
в программе
Torrent suite
Специальные модули
для разных приложений
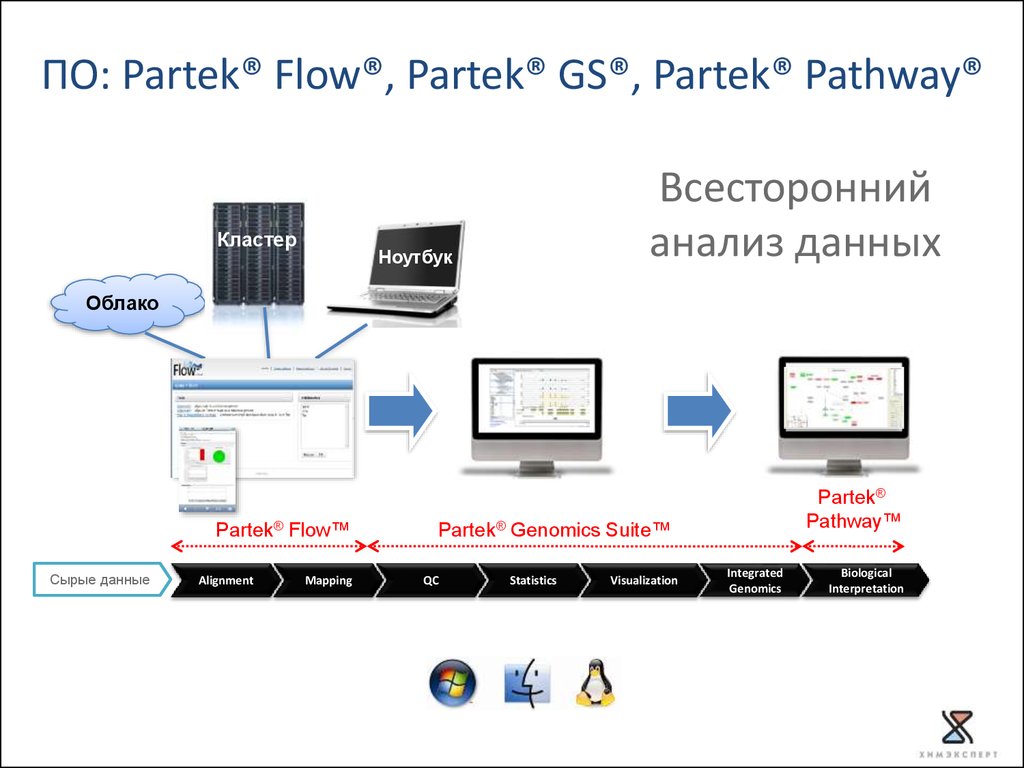
51.
ПО: Partek® Flow®, Partek® GS®, Partek® Pathway®Кластер
Всесторонний
анализ данных
Ноутбук
Облако
Partek® Flow™
Сырые данные
Alignment
Mapping
Partek®
Pathway™
Partek® Genomics Suite™
QC
Copyright © 2011 Partek Incorporated. All rights reserved.
Statistics
Visualization
Integrated
Genomics
Biological
Interpretation
51
52.
Partek® Flow® analysis softwareХарактеристики
Интеграция с Torrent Suite™ Software
Работает в облаке и на кластере
Online доступ к данным в любом месте в любое время
Pre-installed Analysis Pipelines for Ion Torrent™ RNA-Seq Data
AmpliSeq Pipeline
Whole Transcriptome Pipeline
Small RNA-Seq Pipeline
53.
Partek® Flow® analysis softwareКоличественное картирование транскриптома
Обнаружение новых транскриптов
Использование всех доступных баз данных
Оценка представленности отдельных изоформ
Анализ дифференциальной экспрессии
Обнаружение дифференциальной экспрессии
Анализ ген-специфических параметров
Определение сплайс-вариантов
Variant Detection
Обнаружение фьюжн-генов
Обнаружение и аннотация SNPs
Обнаружение SNVs
Классификация (intronic, exonic, UTR, etc.)
Прогнозирование влияния на
аминокислотную последовательность