

")







")


")






")
")
")
")

 aberration (SCA) frequency analysed by FISH in human blood lymphocytes")






 человека")


, donor 51 y. and АТ8SP (11 y.). For purposes of detection of the number of nuclei, the same field of vision is presented, after DAPI coloring.")




, donor 51 y. и АТ8SP (11 y.). DAPI")

, donor 52 y, AT6SP (23 y.) and АТ8SP (11 y.)")

")

 и больной с синдромом Секкеля Sc1SP (вни")






")
")
. Acta Neuropathol Commun. 1:4. doi: 10.1186/2051-59")































")
")
")
")
")





Similar presentations:




Accelerated aging diseases and their genetic causes Complex pattern of aging markers in primary human fibroblasts
1. Accelerated aging diseases and their genetic causes Complex pattern of aging markers in primary human fibroblasts
2.
• Primary non-transformed cell cultures tend tochange with every passage. Population doubling
time increases, cell morphology alteres, larger
rotund cells appear, which are regarded as
older, in comparison with smaller oblong ones
(Мikhelson, 1984; Lorenzini et al., 2005).
Biochemical markers appear simultaneously
with morphological alterations. They
characterize aging cells in culture, defined as
“aging markers”, which include changes in
chromatine, nucleus and cytoplasmatic skeleton,
high level of non-repaired DNA damages, etc.
(Campisi, 2005); as well as in cell instability
towards action of damaging agents, primarily
hydrogen peroxide (Chen et al., 1998, Ryter et
al., 2007).
3. Cells in Culture (5, 10, 15 and 40 p)
4. Cell lines from patients
AT8SPWS(1609)
Cell lines
from patients
HGS1SP
5.
• The most frequently used marker of agingis lysosomal β-galactosidase (SA-β-gal)
associated with aging, its activity
dramatically increasing in old cells (Dimri
et al., 1995; Lee et al., 2006)
6.
• Primary fibroblasts of skin from donors of variosages, and patients with premature ageing with
Hutchinson-Gilford syndrome (children’s
progeria), and the Werner one (adult progeria),
normally serve as model for the detection of
ageing markers
• Fibroblasts taken from old donors, and from the
progeria patients, are currently regarded as
containing more ageing markers, than those
taken from young healthy donors (Scafidi,
Misteli, 2006, Sedelnikova et al., 2008).
• Fibroblasts of skin of other mammals (mice) may
also be applied for the detection of ageing
markers (taking into account the Hyflick limit)
7. Hutchinson-Gilford syndrome
Autosomal dominant,
emerging “de novo”, getting,
probably, from the father site
15 (21)
LMNA 1q21.2
FACE-1/ZMPSTE24 1q34
Skin atrophy.
Bird’s face
Loss of subcutaneous fat
Hair loss.
Arteriosclerosis
Increased metabolic rate
Hypogonadism
Grow hormone insensitivity?
Slow growth.
Reduced replicative life span
of cultured cells
Shot telomeres
DNA repair defects?
Silent mutation in (1-4)-βGalactosyl-transferase gene
(594C>T)
8. Lamine A
9. Nuclear lamina
• Structural transformations of the nuclear lamina,occurring as a result of the accumulation of aberrant
product of the LMNA gene, progerin, were demonstrated
in the course of the study of cells of patients with
Hutchinson-Gilford syndrome, as well as of cell cultures,
acquired from aged donors (Scaffidi, Misteli, 2006).
Results acquired by these authors, taken in complex with
the present-day data concerning participation of
components of nuclear lamina, including lamin А in the
wide range of cell processes. Allow to regard structural
alterations of nuclear lamina as the basic process for
induction of markers of ageing, associated with various
levels of cell regulation .
10. Scaffidi P., Misteli T. 2006. Lamin A-Dependent Nuclear Defects in Human Aging. Science 312 : 1059 – 1063
Rusinol,Sinensky, 2006
Scaffidi P., Misteli T. 2006.
Lamin A-Dependent Nuclear
Defects in Human Aging.
Science 312 : 1059 – 1063
Farnezyl-proteintranspherase
Ras-converting
enzyme or
Zmpste24
S-adenosylmetionin;
isiprenylcarboxymethyltranspherase
Zmpste24
lamine А
«progerin»
11. Lamin A and progerin processing (Ramirez et al., 2007)
12. Chen et al., 2003. LMNA mutations in atypical Werner’s syndrome. Lancet 362 : 440–445.
Ramirez et al., 200613. ZMPSTE24 gene
Ramirez et al., 200614. Nuclear lamina (lamin A/C detection)
• The nuclear defects (Scaffidi,Misteli, 2006; Smirnova et al.,
2008)
• Healthy donor (11 у.) – 5 %
• Healthy donor (87 y.) - 20%
• Atypical Werner’s syndrome –
40%
• Hutchinson-Gilford’s syndrome –
80%
15.
• Accumulation of foci of γ-Н2АХ is observed innuclei of aging cells in culture, cells from old
donors, and from Progeria patients (Sedelnikova
et al., 2004,2008).
• This phenomenon may be linked to both
accumulation of either non-repaired DSBs or,
modifications of chromatine, apprehended as
DSBs by protein kinases АТМ and DNA-PK after
DNA damage (Stiff et al., 2004), or else, ATR
kinase during replication halt (Takahashi, Ohnishi,
2005); as well as by appearance of uncapping
telomeres (Hao et al., 2004).
• As a result, accumulation in cell population of
cells with foci of γ-Н2АХ occurs, which forms an
objective criterion of ageing on cell level.
16. Scaffidi P., Misteli T. 2006. Lamin A-dependent nuclear defects in human aging. Science. 312:1059-1063
17.
• Cells change their epigenetic status with age.Heritable changes in gene regulation with no
changes in the DNA sequence itself are
considered as epigenetic determinants. In recent
years, the role of epigenetic mechanisms in the
process of carcinogenesis (Jones, Baylin, 2002),
as well as in cellular and organismal aging
(Wilson, Jones, 1983; Issa, 2003; Fraga et al.,
2005) was demonstrated.
18. Scaffidi P., Misteli T. 2006. Lamin A-dependent nuclear defects in human aging. Science. 312:1059-1063
19. Werner syndrome
Autosomal recessive
53 (60)
Loss of WRN, a RecQ family helicase.
Skin atrophy.
Hair graying/loss.
Arteriosclerosis
Osteoporosis
Muscle atrophy
Cataracts
Hyperlipidemia
Mild diabetes melitus, type 2
Hypogonadism
Cancer (sarcomas)
Slow growth.
Reduced replicative life span of cultured
cells
Chromosome rearrangements
Sensitivity to 4-NQO and camptothecin
(topoisomerase I poison)
Increased mutation rate, particulary DNA
deletions
Rapid telomere shortening during cellular life
span.
DNA repair defects
DNA replication defects (?)
20. Werner syndrome gene
21. LMNA mutations (Chen et al., 2003. Ramirez et al., 2006)
22.
Bloom syndromeAutosomal recessive
20 (40-50)
BLM-helicase (RecQ-homologue)
Increased metabolic rate
Slow growth.
Diabetes melitus
Hypogonadism
Neurodegeneration
Immunodeficiency
Cancer
Telangiectasias on the face, forming
the red “butterfly”
Low DNA synthesis (2 times) with
the normal rate of DNA-polymerases
α, β, γ
Chromosomes rearrangements
Icreased level of recombination
23. RecQ family helicases (Hickson 2003)
24. RecQ-helicases in DNA repair (Nakoyma 2002)
25. Protein-protein interaction of WRN и BLM helicases (Nakoyma 2002)
)26.
• In the study of cells from a patient withHutchinson-Gilford syndrome, besides the
identification of all markers of aging described
by Scaffidi P., Misteli T (2006), we observed a
significant discrepancy between these cells and
cells from old donors. The level of stable
chromosomal aberrations in the investigated
cells was not elevated and, by this the marker,
normally showing the "real" biological age, our
patient was consistent with her 9 years.
27. The stable chromosome (1, 4, 8 or 12) aberration (SCA) frequency analysed by FISH in human blood lymphocytes
Age dynamics
Age
Number of
respondents
SCA
frequency
(%)
0-19
20-29
30-39
40-49
50-59
60-69
18
38
29
28
24
13
0.15±0.05
0.43±0.04
0.75±0.06
0.89±0.06
1.09±0.08
1.53±0.14
Progeria
syndromes
SCA
frequency (%)
Werner’s syndrome,
Mulde form
0.94±0.38
Atipical Werner’s
syndrome
1.88±0.54
Hutchinson-Gilford’s
syndrome
0.07±0.07
The patients with Werner’s
syndrome demonstrate increased
SCA level, corresponding to
premature aging of organisms.
The lymphocytes from patient with
Hutchinson-Gilford syndrome have
no time for SCA formation
28.
Cutix laxa• Autosomal
recessive
• (40)-50
• Skin atrophy
• DNA repair defects
29. The telomere fragments lenght
1.HeLa cells2.Normal donor
lymphocytes
3.AT2SP (ataxiatelangiectasia patient)
4.Cutis laxa patient
Size in KB
30.
31. Роль АТМ в клеточном ответе на возникновение двунитевых разрывов
32.
Ataxia-telangiectasia (Luis-Bar syndrome)Autosomal recessive
20 (40-50)
ATM, a protein kinase of the PI-3 kinase
family
Neurodegeneration
Immunodeficiency
Cancer (leukemias and lymphomas)
Occulocutaneous telangiectasias
Progeroid skin and hear changes
Hypogonadism
Defectif cell cycle chekpoint arrest
Reduced replicative life span
Chromosomal rearrangements
Sensitivity to ionizing radiation
Inoppropriate apoptosis
Shot telomeres
Delayed/absence P53 induction and
accumulation after DNA damage
Defects in repair of DNA double-strand
breaks
Defects in V(D)J recombination
33. Атаксия-телеангиэктазия АТМ
АТМ
Дефекты репаративного синтеза
ДНК, нарушения клеточного цикла,
высокая
частота
спонтанных
хромосомных
аномалий,
увеличенная чувствительность к
ионизирующим
излучениям
и
радиомиметикам, к УФ-свету и
агентам сходного действия (таким,
как 4-нит-рохинолиноксид)
Появляется у одного из 40 тыс.
новорожденных,
основные
поражения
отмечены
в
нервной и иммунной системах
(мозжечковая атаксия, приводящая к
нарушениям координации мышц,
шатающейся
походке
и
прогрессирующей
умственной
отсталости, кожным нарушениям,
предрасположенности к раковым
заболеваниям и др.)
34. PIKK (phosphatidylinositol 3-kinase-like protein kinases) человека
35. MOSAICS
• Мозаичнаякультура
линий AT1SP
и AT9SP:
детекция
фокусов РАТМ.
• Справа –
DAPI.
36. SA-beta-GAL in AT-cells
37. Detection of γ-Н2АХ in nuclei of human fibroblasts VH-10 (11 y), donor 51 y. and АТ8SP (11 y.). For purposes of detection of the number of nuclei, the same field of vision is presented, after DAPI coloring.
1.08±1.074.06±1.28*
12.32±2.09**
38.
39.
40.
• DNA damage caused by ionizing radiation isaccompanied by ATM phosphorylation of serine
(S1219) in 53BP1 (Lee et al., 2008). This protein also
plays an important role in the checkpoint response
of the cell to damage (Iwabuchi et al., 2008, Eliezer et
al., 2008). The appearance of foci 53ВР1, as well as
foci of γ-Н2AХ can be regarded as one of the reliable
markers of aging cells.
• 53ВР1 protein forms a traditional marker of repair
processes (Wilson, Stern, 2008). Appearance and
elimination of 53ВР1 foci after DNA damage tends to
coincide with γ-Н2АХ dynamics, as they often
colocalize in the zone of DSB
41. 53ВР1 foci in human fibroblast nuclei VH-10 and АТ8SP
• Diffuse coloringtends to occur in
coloring by 53ВР1
antibodies in intact
cells, while clearcut foci form after
DNA damage. Such
foci tend to form in
AT patients’ cells in
many intact cells,
i.e. 16% and 18%
respectedly, while
there are only 6 %
of such cells in
control.
42. Detection of НР1-γ in nuclei of human fibroblasts VH-10 (11 y), donor 51 y. и АТ8SP (11 y.). DAPI
170,71 ± 1,91134,45 ± 4,95*
Fluorescence
intensity
93,01 ± 3,55*
43. SIRT6 SIRT1
44. Detection of 3меК9Н3 in nuclei of human fibroblasts VH-10 (11 y), donor 52 y, AT6SP (23 y.) and АТ8SP (11 y.)
45. 3meH3 fluorescence intensity in human fibroblasts
• In the case of AT8SP, thelevel of trimethyl derivatives
of histone H3 (K9 and K27),
in contrast to the cells from
old donors and other
progeria, is not falling, but
rather rising, as it is shown
in tumor cells.
• In the case of AT6SP, the
level of 3meK9H3 in contrast
to the cells from AT8SP is
falling as in the cells of old
donors, but 3meK27H3 rather
rising, as in AT8SP.
The quantity of 3meK9H3 may
serve as a prognostic marker of
gravity of the disease
Cell strain 3meK9H3
3meK27H3
VH10
(11y.)
47.12±4.36
17.43±1.87
AT6SP
(23 y.)
16.56±1.63
34.12±2.84
AT 8 SP
(11 y.)
153.6±22.99 33.12±2.07
52 y.
26.4±5.01
10.22±1.36
46. Seckel syndrome (atr)
• O'Driscoll M, Ruiz-Perez VL, Woods CG,Jeggo PA, Goodship JA. 2003. A
splicing mutation affecting expression
of ataxia-telangiectasia and Rad3related protein (ATR) results in Seckel
syndrome. Nat Genet.,33:497-501
47. Синдром Секкеля
• Микроцефалия,Умственная
отсталость
• Карликовость
• Задержки развития
Диспластический
фенотип.
• Microcephalic
primordial dwarfism
(MPD)
48. Совместная окраска антителами к рАТМ и активным киназным доменам АТМ/АTR здорового донора VH-10 (вверху) и больной с синдромом Секкеля Sc1SP (вни
Совместная окраска антителами к рАТМ и активнымкиназным доменам АТМ/АTR здорового донора VH10 (вверху) и больной с синдромом Секкеля Sc1SP
(внизу)
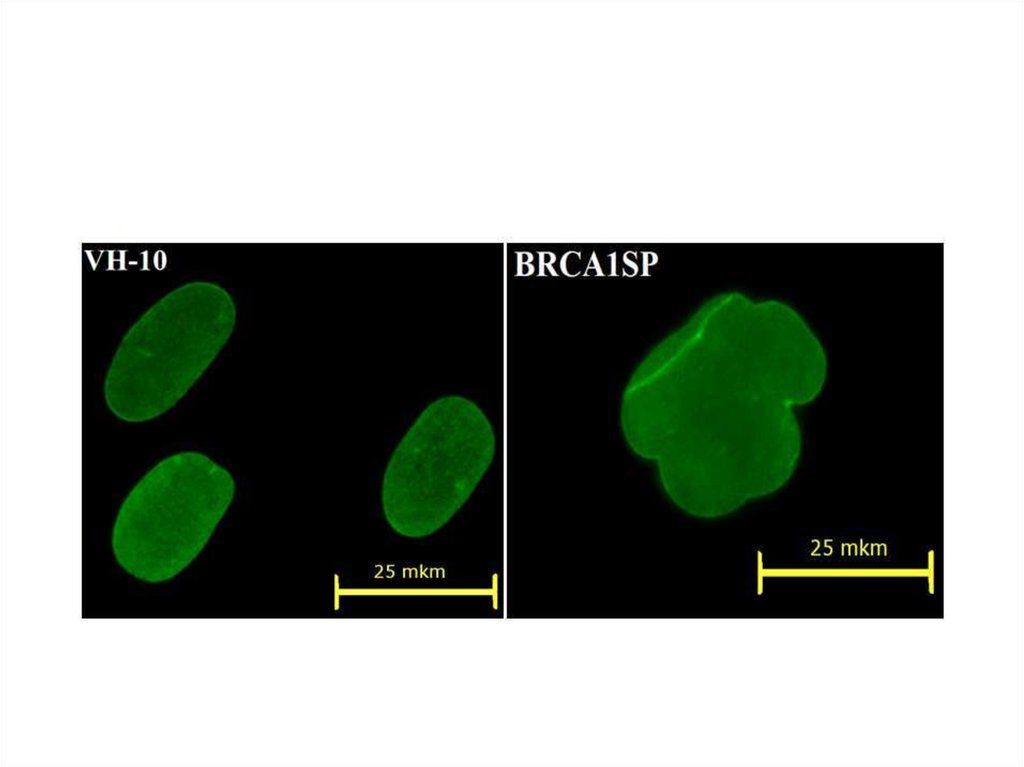
49. Ss1SP BRCA1SP
50. Ядерная оболочка, окраска антителами к ламину А\С
51.
52.
53. Mutations in TRAIP cause primordial dwarfism
Mutations in TRAIP cause primordial dwarfismHarley et al., 2016.
TRAIP promotes
DNA damage
response during
genome replication
and is mutated in
primordial
dwarfism.
Nat Genet.
48(1):36-43
The TRAF [tumor
necrosis factor
(TNF) receptorassociated factors]
-interacting protein
(TRAIP) functions
as (RING)-type E3
ubiquitin ligase, but
its physiological
substrates are not
yet known.
54. TRAIP localizes to sites of UV-induced DNA damage
TRAIP localizes to sites of UVinduced DNA damage
(a) TRAIP localizes to DNA damage sites
induced by UV laser microirradiation both in
the absence and presence of pre-treatment
with BrdU as a damage sensitizer.
Representative images, before and after UV
laser microirradiation. Scale bar, 5 μm. (b)
GFP-TRAIP colocalizes with γH2AX and
with RFP-PCNA at sites of UV laser-induced
damage. Representative images of UV
laser-irradiated GFP-TRAIP expressing cells
immunostained for γH2AX (pre-sensitized
with BrdU) or co-expressing RFP-PCNA (no
BrdU pre-treatment) as indicated. Scale bar,
5 μm. (c, d) GFP-TRAIP is detected by a
Proximity Ligation Assay (PLA) in close
proximity to PCNA, an association enhanced
after UV-induced damage. (c)
Representative images of PLA
signals/nucleus in doxycycline-inducible
GFP-TRAIP HeLa cells before and after
damage with 25 J/m2 UV-C. Scale bar, 5
μm. (d) Quantification of PLA
signals/nucleus. Box plots, center line
denote mean values, box 25/75 %, whiskers
5/95 %, data pooled from n=2 independent
experiments, n>65 data points per condition
per experiment; Mann Whitney rank sum
test: *** p<0.001. (e) TRAIP accumulates at
sites of localized UV damage, colocalising
with RPA and γH2AX. Representative
immunofluorescence images of MRC5 cells
transfected with GFP-TRAIP or GFP alone
after UV-C irradiation through 3 μm
microfilters. Scale bar, 5 μm.
55. PCNA-mutations
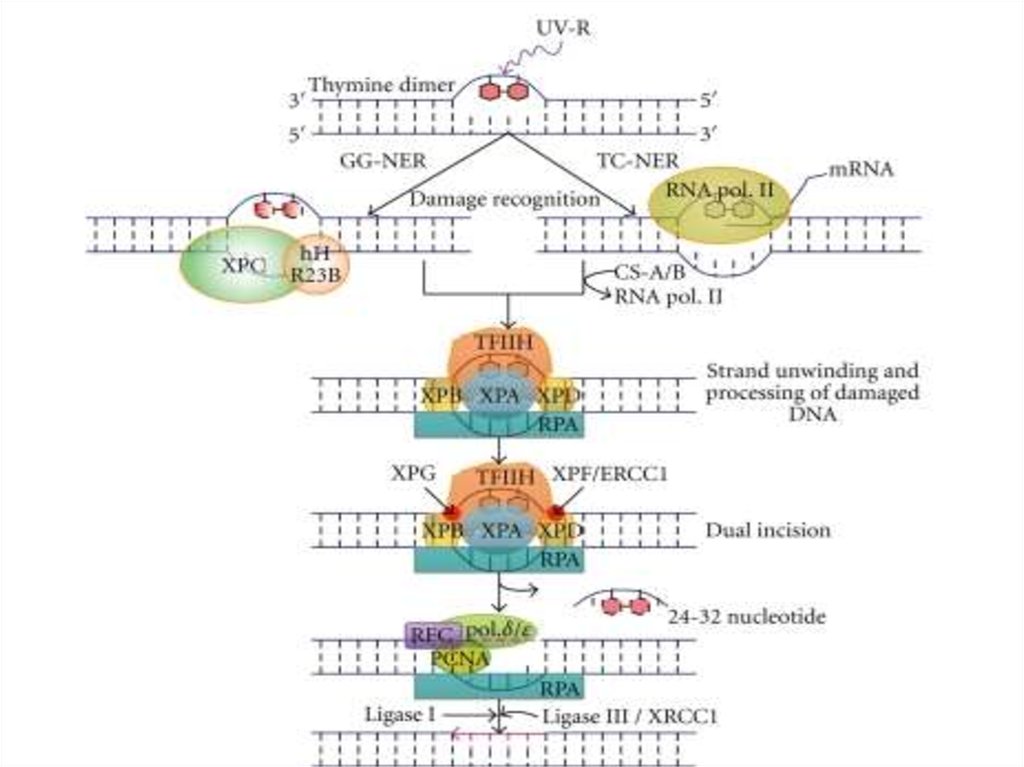
56. Excision repair systems (Spivak G., 2004)
57.
58. Xeroderma pigmentosum (XP)
• XPA, XPB, XPD, XPC, XPE, XPF, XPG ,• XP-V не выявлен дефект NER
• Дефекты эксцизионной репарации
нуклеотидов
(нарушение
вырезания, застройки брешей и др.).
Разнообразные
дефекты
репарационных процессов.
• При
XP-вариант
наблюдается
изменение параметров репликации
ДНК
• Сверхчувствительность к УФ-свету,
• ведущая к появлению красных
пятен на коже, переходящих в
незаживающие коросты и нередко в
рак
кожи;
неврологические
расстройства);
поражения
век,
бровей и глаз. Распространение: 1
случай на 250000 человек в Европе
и США; 1 :40000 человек в Японии
59. Lai et al., 2013. The influence of DNA repair on neurological degeneration, cachexia, skin cancer and internal neoplasms: autopsy report of four xeroderma pigmentosum patients (XP-A, XP-C and XP-D). Acta Neuropathol Commun. 1:4. doi: 10.1186/2051-59
Lai et al., 2013. The influence of DNA repair on neurological degeneration,cachexia, skin cancer and internal neoplasms: autopsy report of four xeroderma
pigmentosum patients (XP-A, XP-C and XP-D). Acta Neuropathol Commun. 1:4.
doi: 10.1186/2051-5960-1-4.
XP patients studied. A and B: Case 1 XPA patient (A) at age 17y with numerous
freckle-like pigmented lesions on sun
exposed skin and (B) at 37y with marked
cachexia and thinning of subcutaneous
tissues of face and chest. She had more that
100 surgical procedures on her face for
removal of skin lesions. C: Case 2 XP-D
patient at age 40y. She had been well
protected from sun exposure since early
childhood and had only few pigmented
lesions and skin cancers. D: Case 3 XP-C
patient, at age 29y with multiple freckle-like
pigmented lesion on sun exposed skin and
cheilitis. The patient underwent many
surgical procedures for removal of skin
cancers on her face. E and F: Case 4 XP-C
patient, at age 28y (E) with multiple
pigmented lesions, telangiectasia, cheilitis
and corneal clouding. Multiple surgical
procedures were performed on her face for
removal of skin cancers and at age 48y (F)
following exenteration of both orbits for
treatment of ocular squamous cell
carcinomas.
60.
Cockayne syndrome (CS)Autosomal recessive
CSA (WD-repeat protein, β-subunit GTP
protein homologue)
CSB (ATPase of Swi2 family),
XPB and XPD (helicases in TFII-H),
XPG (yeast Rad2 homologue,
endonuclease)
20 (40?)
Bird’s face
Loss of subcutaneous fat
Skin photosensitivity
Neurodegeneration
Hypogonadism
UV-sensitivity
Impaired transcription-coupled nucleotide
excision repair
Decreased recovery of transcription after
irradiation
General RNA-polymerase II transcription
defects
61. TTD
62. Анемия Фанкони
• FAA, FAB, FAC, FAD, FAE, FAF, FAG -19 группкомплементации
• Дефекты репарации повреждений от химических
мутагенов
и
канцерогенов
(по
не
УФ-свста),
обусловленные дефектами эндонуклеаз, дефектами
распознавания кросс-сшивок ДНК;
• пониженная
способность
к
апоптозу
после
ионизирующего облучения; двукратное удлинение G2фазы клеточного цикла
Сверхчувствительность к химическим мутагенам и
канцерогенам, уменьшение количества всех клеточных
элементов крови, различные аномалии врожденных
способностей, деформация пальцев и другие виды
скелетных нарушений, урогенитальные нарушения,
микроцефалия, микрофтальмия, дефекты уха и потеря
слуха, сердечные и гастроинтестинальные нарушения
63.
• Hum Genomics. 2015 Nov 24;9(1):32. doi: 10.1186/s40246-015-0054-y.• Update of the human and mouse Fanconi anemia genes.
• Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V.
• Fanconi anemia (FA) is a recessively inherited disease manifesting
developmental abnormalities, bone marrow failure, and increased risk of
malignancies. Whereas FA has been studied for nearly 90 years, only in
the last 20 years have increasing numbers of genes been implicated in the
pathogenesis associated with this genetic disease. To date, 19 genes have
been identified that encode Fanconi anemia complementation group
proteins, all of which are named or aliased, using the root symbol
"FANC." Fanconi anemia subtype (FANC) proteins function in a common
DNA repair pathway called "the FA pathway," which is essential for
maintaining genomic integrity. The various FANC mutant proteins
contribute to distinct steps associated with FA pathogenesis. Herein, we
provide a review update of the 19 human FANC and their mouse orthologs,
an evolutionary perspective on the FANC genes, and the functional
significance of the FA DNA repair pathway in association with clinical
disorders. This is an example of a set of genes--known to exist in
vertebrates, invertebrates, plants, and yeast--that are grouped together on
the basis of shared biochemical and physiological functions, rather than
evolutionary phylogeny, and have been named on this basis by the HUGO
Gene Nomenclature Committee (HGNC).
64. Simplified model of the Fanconi anemia pathway
• . Activation of FANCD2 and FANCI by the FA core complexvia monoubiquitination (orange circles) regulates
downstream genes involved in recombination repair of DNA
crosslinks.
65.
66.
67. Age of onset of SCC in FA patients with and without HSCT
• Forty-three of 83 female FA patients (51.8%) and 17 of 46 male FApatients (37.0%) developed SCC (average age at SCC diagnosis
for 48 FA patients without HSCT 30.0 years, for 12 FA patients after
HSCT 25.8 years).
68. Telomeres
• Telomeres, being the final fragments of eukaryotic chromosomes,form one of the most widely studied at present time in the framework
of study of primary mechanisms of the organism ageing, potential
factors, conditioning life span. Considerable interest directed at
these specialized complexes, is conditioned by their unique
functions in the securing of the cell genome (Blackburn, 2001). Apart
from preventing the chromosome fusion, telomeres are responsible
for their fixation at cell membrane (Podgornaya et al., 2000; Hediger
et al., 2002; Rose et al., 2004), for the mitotic and meiotic
segregation of chromosomes (Conrad et al.,1997; Kirk et al., 1997;
Dynek, Smith, 2004), and for their meiotic coupling (Rockmill,
Roeder, 1998), for the stabilization of broken chromosomes (Jager,
Philippsen, 1989; Pennaneach et al., 2006), and for their defence
against reparation systems (Shay, Wright, 2007; Mirsi et al., 2008),
as well as being responsible for gene expression (Baur et al., 2001;
Pedham et al., 2006).
69. Age-dependent telomere length
70. Telomere length
71. Factor analysis of mean telomere length, genetic polymorphism, and age, by senior age respondents
• Regular correlationbetween telomere length
with respondent age
appears after having
attained certain age,
when adequate strategy
of active longevity has
been attained, and
individual differences may
be traced back.
Data
Factor 1
Factor 2
Age
-0,01
0,72
АСЕ
0,64
-0,13
5НТR2А
0,78
0,08
5НТТ
0,31
0,43
Telomere
length
0.26
-0.65
Expl.Var
1,182912
1,140631
Prp.Totl
0,286582
0,278126
72.
Запретить73.
74. Комплекс генов анемии Фанкони
75. Simplified model of the Fanconi anemia pathway
• . Activation of FANCD2 and FANCI by the FA core complexvia monoubiquitination (orange circles) regulates
downstream genes involved in recombination repair of DNA
crosslinks.
76. Mechanism of ICL repair in the FA pathway on collision of a replication fork with an ICL
Анемия Фанкони (A, B, C, D1, D2, E, F,
G, I, K, L, M, N, O, P, and Q). Эти белки
делятся на 3 группы: (1) белки
корового комплекса; (2) FANCI и
FANCD2 белки, составляющие
комплекс ID2 и (3) эффекторные белки
(A) FA активируется во время фазы S
при обнаружении ICLs или сходных
повреждений ДНК. Коровый комплекс
FA привлекается в зону повреждения
MHF1-MHF2-FANCM комплексом.
(B) Комплекс ID2
моноубиквитинируется и связывается с
зоной повреждения ДНК. Комплекс BL-100 способствует этому
убиквитинированию совместно с еще
двумя субкомплексами (A-G-20 and CE-F)
(C) Нуклеазы XPF/FANCQ-ERCC1
совместно с SLX4/FANCP, incise the
Dпроводят ДНК-инцизию
(D) Моноубиквитинированный
комплекс ID2 привлекает белки
репарации, включая BRCA1,
BRCA2/FANCD1, FANCJ,
PALB2/FANCN, and RAD51C/FANCO.
(E) После успешной репарации
комплекс, ID2 деубиквитинизируется
USP1-UAF1 , что способствует его
высвобождению из хроматина.
77. FA/BRCA pathway and crosstalk between FA and other DNA repair pathways
В ответ на сигнал о повреждении ДНК (фосфорилирование ATR/ATM),через FA/BRCA патвэй [1], формируется
коровый комплекс FA – состоящий из FANCA (A), FANCB (B), FANCC (C), FANCE (E), FANCF (F), FANCG (G), FANCM
(M), и FANCL (L) белков плюс FAAP20, FAAP24, и FAAP100 (FAAP). Этот комплекс связывается UBE2T(T) через
FANCL, моноубиквитинируя и активируя димер FANCD2/I. FANCD2/I (D2/I) переносится в зону повреждения и
привлекает туда эе белки-эффекторы, включая BRCA1 (S), BRCA2 (D1), RAD51 (R), BRIP1 (J), PALB2 (N), RAD51C
(O), SLX4 (P), and ERCC4 (Q), и ждругие репарационные факторы (FAN1). Через FANCM/BS патвэй [2], FA коровый
комплекс связывается с комплексом BS путем взаимодействия FANCM-RMI1и TopoIIIα с BS, также привлекая его в
район повреждения. Через FANCD2/ATM патвэй [3],, FANCD2 фосфорилируется ATM и колоколизуется с комплексом
NMR , что вызывает S-арест. complex
78. Overview of FA pathway genes identified in eukaryotic lineages.
Representative species include mammals (Homo sapiens, Mus musculus, and Gallus gallus), amphibian (African clawed toad, Xenopus
laevis), fish (zebrafish, Danio rerio), sea squirt (Ciona intestinalis), insect (Drosophila melanogaster), worm (Caenorhabditis elegans), yeast
(Saccharomyces cerevisiae), and plant (Arabidopsis thaliana). FANC genes are grouped into three classes. Group I includes nine genes that
encode proteins that form the FA core complex; group II encodes FANCD2 and FANCI that form the D2/I complex; group III comprises eight
genes that encode FA effector proteins that function downstream of D2/I complex. Lower eukaryotes tend to be missing orthologues of the FA
core complex
genes. A = FANCA, B = FANCB, C = FANCC, D2 = FANCD2, E = FANCE, F = FANCF, G = FANCG, I = FANCI, L = FANCL, M = FANCM, D1 =
BRCA2/FANCD1, J = BRIP1/FANCJ, N = PALB2/FANCN, O = RAD51C/FANCO, P = SLX4/FANCP, Q = ERCC4/FANCQ/XPF, R = RAD51/FA
NCR, S = BRCA1/FANCS, T = UBE2T/FANCT. If we extend this gene family update to include prokaryotes, it might be noted that, whereas no
orthologs of any of the 19 eukaryotic FANC genes exist in prokaryote genomes, RAD51 (as a nineteenth FANC member in living organisms)
qualifies as a homologue of bacterial RecA
79. Domain architecture and structure of FANCD2 and FANCI
. (A) Schematic of the FANCD2 protein
indicating the amino-terminal NLS (nuclear
localization signal) domain (green), CUE
(coupling of ubiquitin conjugation to
endoplasmic reticulum degradation) domain
(maroon), PIP-box (PCNA-interacting
protein motif) (orange), and the C-terminal
EDGE motif (purple). Functionallycharacterized phosphorylation sites (teal)
and K561 monoubiquitination site (yellow)
are indicated by small circles. (B) Schematic
of the FANCI protein indicating the Leu
(leucine zipper) domain (light blue), ARM
(armadillo repeat) domain (pink), and Cterminal EDGE motif (purple) and NLS
domain (green). The S/TQ motif (teal) and
K523 site of monoubiquitination (yellow) are
indicated by small circles. (C) Mouse FanciFancd2 heterodimer crystal structure
represented as both surface and ribbons
with domains indicated. This structure was
solved by the Pavletich group in 2011(PDB
ID: 3S4W).
80. Comparison of the SCF multi-subunit ubiquitin ligase protein complex and the FA core complex
. (A) The Skp1/Cullin/F-box protein (SCF) complex includes the Cullin protein, which
acts as a scaffold to bridge the catalytic E3 ubiquitin ligase RBX1 to the adaptor
protein Skp1, and the F-box protein. The F-box protein recognizes and recruits the
target protein for ubiquitination by the E2 ubiquitin-conjugating enzyme, UBC. (B) We
propose that the FANCA protein is structurally analogous to Cullin, and may link the
E3 ubiquitin ligase FANCL with the putative adaptor protein FANCC. FANCC has
been shown to interact with both FANCA and FANCE, indicating that it may function
analogously to Skp1. FANCE may be analogous to the F-box protein. FANCE is
known to interact directly with FANCD2 and may facilitate its monoubiquitination of
FANCL and UBE2T.
81. Models for FANCD2 and FANCI monoubiquitination
The schematics depict several potential outcomes upon monoubiquitination of FANCD2 and FANCI, which would
preclude further ubiquitination. (A) The ID2 heterodimer inactivation model. Following monoubiquitination, ID2
heterodimerization occurs and is stabilized through a noncovalent interaction between monoubiquitin covalently linked
to FANCI K523 and the FANCD2 CUE domain. There is also possibly a reciprocal interaction between
monoubiquitinated FANCD2 K561 and an UBD in the carboxy-terminus of FANCI, shielding FANCD2 from further
ubiquitination. (B) The FANCD2 self-inactivation model. Monoubiquitination could promote an intramolecular
association between ubiquitin covalently attached to K561 and the amino-terminal CUE domain, resulting in a closed
conformation. (C) The E3 ubiquitin ligase dissociation model. Once FANCL is autoubiquitinated, the ubiquitin moiety
may interact noncovalently with the CUE domain on FANCD2 enabling monoubiquitination of FANCD2 on K561. This
interaction is predicted to be weak and short-lived, leading to rapid dissociation of FANCL and FANCD2, precluding
further ubiquitination.
82. Теломерные белки у человека
83.
• Data acquired in the course of study of primaryfibroblasts from humans, are less contradictory,
and provide a picture which is much more
stable. It is necessary to assess more accurately
the character of dispersion of all markers studied
by decades, and to form a reliable scale.
• It is just this observation which demonstrates
that progeria is a deeply pathological aging and
its analogy with the natural aging is limited.
84. Структурная организация белка Р53
85. SA-β-gal в первичных фибробластах мышей SHR
86. G1 чекпойнт
87. S-чекпойнт
88. G2-чекпойнт
89. Внутриядерная локализация белков во время ответа клетки на повреждение ДНК
90. Механизмы G2-ареста
• АТМ активирует СНК2 через фосфорелированиетриптофана в 68 положении, которая в свою очередь
фосфорелирует серин в 215 положении у CDC25C, что
приводит к блокированию ее функций.
Фосфорелированная форма CDC25C связывается с
белком 14-3-3σ, что поддерживает еe каталитическую
неактивность и способствует переходу в цитоплазму и
секвестрированию.
• Вторая ветвь G2-чекпойнта опосредуется через
ATR/CHK1 активацию. При этом пути одновременно
фосфорелируется-выключается белок CDC25А, а также
фосфорелируется серин-549 белка Wee1
(пртеинкиназа), что облегчает его связывание с тем же
белком 14-3-3σ и приводит к усилению ингибиторной
активности киназ по отношению к CDC2(CDK1). Это
придает второй ветви большую гибкость в контроле и
консолидации G2-ареста.
91. Механизмы G2-ареста(2)
• После облучения резко падает уровеньмРНК циклина В, возможно из-за ее
повышенной нестабильности, причем этот
эффект определяет протяженность G2ареста.
• Циклин В во время G1 и S фаз имеет
цитоплазматическую локализацию и
перемещается в ядро только к началу
митоза. Белок 14-3-3σ приводит к
секвестрированию циклина В в цитоплазме
в ответ на повреждение ДНК.
92. Механизмы G2-ареста(3)
• PLK1 и PLK3 (Polo-like kinase). Белки этогосемейства принимают активное участие в
митозе, включая вход и выход из него.
PLK1 является позитивным регулятором
CDC25C-активности в необлученных
клетках и, специфически фосфорелируя
ее, способствует вхождению в митоз.
PLK3, напротив, активируется АТМ в ответ
на повреждение ДНК, взаимодействует с
CDC25C, фосфорелируя ее по серину-216,
что приводит к ингибированию ее
активности.
93. Механизмы G2-ареста(4)
• Остановить вход в митоз при наличииповреждений в ДНК может
взаимодействие PCNA c Р21, CDC25C, и
CDC2(Cdk1)-циклин В, но не
одновременное, а последовательное.
Связывание Р21 и CDC25C с комплексом
PCNA-CDC2-циклин В является
совершенно особым и не позволяет
CDC25C дефосфорелировать CDК1 для
активации митоза. Р21 может блокировать
САК, которая активирует CDК1 путем
фосфорелирования триптофана в 161
положении.
94. Механизмы G2-ареста(5)
• Роль Р53 в поддержании G2-ареста состоит втом, что он активирует транскрипцию трех
вовлеченных в него белков: GADD45, P21 и 143-3σ и подавляет транскрипцию CDC2(CDK1) и
циклина В.
• Есть данные о вовлеченности в G2-арест
BRCA1, опосредованно через ATM/ATR или
напрямую, через активацию CHK1, но механизм
этого остается неясным.
• G2-реакция клетки на облучение зависит от
фазы цикла, в которую это произошло.
95. Механизмы G2-ареста(6)
• G2 чекпойнт-ответ разделяется на дваразличных пути. Один начинается сразу же
после облучения, захватывает клетки,
облученные непосредственно в G2-фазе и
является АТМ-зависимым, проходящим и
независимым от дозы. Он приводит к резкому
снижению митотического индекса.
• Второй, который развивается позже, в клетках,
облученных на более ранних стадиях
клеточного цикла, является АТМ-независимым,
зато зависимым от дозы и приводит к
накоплению клеток в фазе G2.
96. Репарация, спаренная с транскрипцией. TTD
Повышеннаяфоточувствительность
ДНК
(примерно в 50% случаев
заболеваний),
нарушение
вырезания димеров тbмина или
недостаточная
скорость
застройки
брешей
после
вырезания
нуклеотидов,
возможны
нарушения
транскрипции
Нехватка серы в белках волос
и их луковиц, ведущая к
ломкости волос, "тигровость"
волос (чередование светлых и
темных полос по длине волоса,
выявляемое под микроскопом);
ихтиоз; часто умственная и
физическая
отсталость;
дефекты полового развития;
аномалии
кожи
и
зубов;
97. Репарация межнитевых сшивок
Схема процеса репарации
сшивок у прокариот
соответствует тем же
системам репарации - NER и
HR