









")






























")




")

 (синдром ПАТАУ)")








 - учение о наследственном здоровье человека и путях")
")


 biology
biologySimilar presentations:










Закономерности наследования признаков. Генетика человека. Структурные уровни организации наследственного материала
1.
Закономерностинаследования
признаков.
Генетика человека.
1. Структурные уровни организации наследственного
материала
2. Регуляция экспрессии генов
3. Наследственно обусловленные различия людей
4. Нарушения кариотипа и их фенотипическое
проявление
5. Генетический мозаицизм
6. Структурные аномалии хромосом
1
2. Наследственность-
Наследственностьсвойство организмов обеспечиватьматериальную и функциональную
преемственность
между
поколениями
Наследственность
обусловливает
специфичность
онтогенеза
в
определенных условиях внешней
среды
Наследование – процесс передачи
наследственной информации между
2
поколениями
3. Термин «Плазмида» предложил Дж. Леденберг в 1952г
34.
Карта F- фактора (фактор фертильности=размножения) и R- фактора (фактор
устойчивости к лекарственным средствам)
4
5.
Амплификация – увеличение количества копийгена в данной клетке
5
6.
Генная инженерия – создание организма сзаранее известными свойствами. Это стало
возможным при разработке методов переноса
генетической информации из одного организма в
другой.
Плазмиды бактерий
6
7. Ген
- участок молекулы ДНК, несущийинформацию
о
структуре
полипептидной
цепи
или
макромолекулы.
• Термин ≪ген≫ был предложен в 1909 г.
датским
ботаником
Вильгельмом
Йогансеном.
7
8. Молекула ДНК в клетке полифункциональна, в связи с этим в ДНК имеются последовательности которые:
-контролируют экспрессию генов-контролируют репликацию
-включают и выключают другие гены
8
9. Структурные гены
Последовательностинуклеотидов
(гены),
которые
содержат
информацию
о
структуре
полипептидной цепи, а в конечном
счете
о
структурном
белке
(ферменты, антитела, рецепторные
белки
…..)
называются
структурными генами
9
10. Регуляторные гены
Последовательностинуклеотидов
(гены), которые определяют время,
место и длительность включения
структурных
генов
называются
регуляторными генами
10
11. Система оперона (Ф.Жакоб,Ж.Моно,1961 г)
лактозный оперон кишечной палочки11
12. Может быть 2 состояния оперона:
А) оперон включен.• Лактоза поступает в клетку и соединяется с белкомрепрессором, отсоединяя его от оператора.
• Операторный участок разблокирован, поэтому РНКполимераза через него проходит и осуществляет
транскрипцию с этих структурных генов.
• Образуется иРНК, а затем белки ферменты,
расщепляющие лактозу.
Б) оперон выключен.
• При отсутствии метаболита белок репрессор
соединяется с оператором, блокируя транскрипцию.
• Промотор регулирует РНК-полимеразу.
• РНК-полимераза не может двигаться и не идет
транскрипция.
12
13. Арчибальд Геррод, 1902 г
• Впервые связь между генами и ихконечными продуктами (белками) была
обнаружена английским врачом А. Гэрродом
в 1902 году
• Алкаптонурия (фенотипический признакмоча цвета красного вина). По мнению
Гэррода это наследственное заболевание
обусловлено нарушением азотистого
обмена, в результате которого вместо
мочевины в моче содержится какое-то
вещество темного цвета.
13
14. А.Гэррод и Бэтсон, 1908 г
• больные алкаптонурией являютсярецессивными гомозиготами.
• «По вине» рецессивного гена у них не
происходит какой-то ферментативной
метаболической реакции. Отсутствие
этой реакции, в свою очередь, приводит
к накоплению и выведению субстрата,
который в норме разрушался бы в
результате этой реакции.
14
15.
Гомогентизиновая кислота ->->X-> малеин-ацетат -> Н 0 + С0
2
2
Оксидаза гомогентизиновой кислоты
(отсутствует!)
накапливается
и переходит в мочу
Гомогентизиновая кислота у больных
алкаптонурией на воздухе окисляется и
чернеет, а моча приобретает цвет
красного вина.
15
16. Бидл, Татум, 1940 г
Гипотеза «Один ген - один фермент»Ограничения
- не все белки выполняют ферментативную функцию
- не все белки состоят из одной полипептидной цепи
(олигомерные)
- многие белки имеют четвертичную структуру
Современная трактовка
«Один ген-одна полипептидная цепь»
16
17. Кол-во ДНК в хромосомном наборе человека
На гаплоидный набор приходится 3х109 п.н(пар нуклеотидов)
длина около 1 м
На одну хромосому приходится в среднем 1,2х108 п.н.
длина около 4 см
ДНК в метафазной хромосоме
компактизована в 104 раз по сравнению с
интерфазной
17
18.
Уровни упаковки генетического материала:18
19.
1920.
2021.
2122. Схема различных уровней компактизации хроматина
1. нуклеосома2. нуклеомер, «сверхбусина» Гистоновые и негистоновые белки
3. хромомер
принимают участие:
4. хромонема
5. хромосома
1)в формировании
структуры хромосом
2) в регуляции экспрессии генов
22
23.
• Экзон — последовательность ДНК, котораяпредставлена в зрелой РНК (информативный
участок). В составе гена должен присутствовать
как минимум один экзон.
• Интрон
—
последовательность
ДНК,
включённая между экзонами, не входит в
состав
зрелой
РНК.
Интроны
имеют
определённые
нуклеотидные
последовательности,
обусловливающие
их
границы с экзонами: на 5’-конце — GU
последовательность, на 3’-конце — AG.
• Интроны содержат регуляторные элементы
экспрессии гена и могут кодировать регуляторные
РНК (например, рРНК, тРНК, малые ядерные
23
РНК, короткие интерферирующие РНК)
24.
• Фактор инициации транскрипциивходит в состав первого экзона.
• РНК-полимераза связывается с
промотором в точке инициации,
начинается транскрипция переписывание
порядка
положения
нуклеотидов
в
молекуле ДНК.
• Элонгация
РНК-полимераза
движется по ДНК-матрице в
направлении 3’ → 5’. Синтез РНК
протекает
по
принципу
комплементарности.
Терминация - синтез про-мРНК
прекращается
в
точке
терминации последнего экзона.
24
25.
Процессинг и сплайсинг мРНК• Процессинг
–
совокупность
реакций, в результате которых из
про-м-РНК
вырезаются
неинформативные
участки
(интроны)
и
остаются
информативные участки (экзоны).
• Сплайсинг (от англ. splice —
≪соединять
концы
чеголибо≫) - процесс объединения
экзонов друг с другом.
• Зрелая
мРНК
состоит
из
экзонов, содержит 5’- и 3’нетранслируемые концы. 5’нетранслируемый
конец
кэпирован, 3’-нетранслируемый
конец защищён полиадениновым
25
≪хвостом≫.
26.
Зачем в природе возникла разорванная структураэукариотических генов = зачем природе
понадобилось вводить сложный процесс
сплайсинга и процессинга, включающего разрывы
и соединения концов РНК и уничтожение ¾
синтезированной пре-мРНК?.
Гипотеза: «сплайсинг, с его способностью объединять
разъединенные участки в один ген, может играть важную
роль в эволюции живых организмов (может объединять
разные гены в один, следовательно разных полипептидных
цепей в одну. Таким путем легко могут возникнуть новые
гены).
ДНК-полимераза 1 представлена 1 полипептидной
цепью, но состоит из разных ферментов: собственно ДНКполимеразы и эндонуклеазы. Два домена образуют 2
компактные частицы, связанные коротким полипептидным
мостиком.
26
27.
Транспортная РНК27
28. Рибосомный цикл синтеза белка
Трансляция(от
лат.
translatio
—
≪перенос≫,
≪перемещение≫;
≪перевод≫—
преобразование
последовательности
нуклеотидов
мРНК
в
последовательность аминокислот (синтез белка) на
рибосомах с помощью тРНК.
29.
Современная теория гена1. Ген-часть молекулы ДНК, являющаяся функциональной
единицей наследственной информации
2. Ген занимает определенный участок (локус=цистрон) в
хромосоме
3. Внутри гена могут происходить рекомбинации
4. ДНК, входящая в состав гена, способна к репарации. Не
репарируемые повреждения приводят к мутациям.
5. Существуют гены регуляторные и структурные (модификаторы
и другие)
6. Расположение триплетов (кодонов=сайтов) в структурных
генах колиниарно аминокислотам полипептидной цепи,
кодируемой данным геном (может быть сдвиг рамки
считывания)
7. Генотип, будучи дискретным (состоящим из отдельных генов),
функционирует как единое целое
8. Генетический код универсален
9. Генетический код вырожденный (для многих аминокислот
существует более одного кодона)
10. Гены располагаются в хромосоме в линейном порядке и
образуют группу сцепления.
29
30.
6-8 %~ 90% болезней с
наследственным
предрасположением
Травмы,
ожоги
30
31. Частные разделы генетики человека
Генетикаповедения
Цитогенетика
Гибридизация
клеток
Популяционная
генетика
человека
Евгеника
Генетика
человека
Биохимическая
генетика
человека
Наблюдение,
статистический анализ
семейной
распространенности
нормальных признаков,
пороков развития,
заболевания
31
32. В России ежегодно рождается 40-50 на 1000 детей с врожденной наследственной патологией
• 40% ранней младенческой смертности иинвалидизации
обусловлено
наследственными факторами;
• 30%
коек
в
детских
лечебных
учреждениях
занимают
дети
с
наследственной патологией:
• более половины из них погибают очень
рано
или
имеют
хронические
заболевания.
32
33.
• Наследственныеболезни
встречаются
в
практической
деятельности
врача
любой
специальности т.к.
• у 11-16% больных, поступающих в
педиатрические отделения клиник
диагностируют
генетические
заболевания.
33
34.
• 8,5% детей умирают от заболеваний,связанных с генными мутациями;
• 2,5% детей умирают от заболеваний,
связанных с хромосомными нарушениями;
• 31% детей умирают от так называемой
отягощенности
наследственными
признаками.
• Итого 42%
• 17% детей умирают от заболеваний
неизвестной природы.
34
35. Большой процент больных с наследственной патологией:
• в гематологических клиниках;• 16% в детской нефрологии;
• 50%
детской
слепоты
составляют
наследственные аномалии;
• 50%
больных с нарушенным слухомнаследственная патология;
• Тяжелые формы наследственной патологии
составляют 1-2%. На 5 млн живорожденных
приходится 50-100 тысяч детей с тяжелой
врожденной и наследственной патологией.
35
36.
МАККЬЮСИК 2005 годВсего описано 6 678 синдромов и
признаков у человека среди них –
4 458 - аутосомно-доминантных
1 730 - аутосомно-рецессивных
412 - x – сцепленных
19 - у – сцепленных
66 – митохондриально-наследуемых
36
37. Кариотип
-диплоидный набор хромосом,характеризующийся совокупностью
признаков: число,
размер,
форма,
особенности строения
Постоянство кариотипа поддерживается
механизмами мейоза и митоза
37
38.
В зависимости от расположенияцентромеры хромосомы делятся на типы
(МакКьюсик, 1967).
Типы метафазных хромосом
Концевые спутники (вторичные перетяжки) имеют 13, 14, 15, 21, 22
38
хромосомы
39.
В Денвере в 1960 году была предложенаклассификация хромосом
Идиограмма – графическое изображение
хромосом в порядке убывания их размеров
39
40.
Парижская классификация хромосом (1970)Идиограмма
Х хромосома
Метафазная пластинка
FISH-окраска
У хромосома
40
41.
• В 1971 году предложена система символов,кратко описывающая кариотип человека:
• 46,ХХ – кариотип здоровой женщины
• 46,ХY – кариотип здорового мужчины
Во время мейоза возможны нерасхождения
хромосом, тогда в одну гамету попадает 24
41
хромосомы, в другую – 22.
42. Синдром Клайнфельтера (47, ХХУ)
Характерные симптомы:высокий рост, длинные ноги
недоразвитие половых органов
(яички уменьшены в размерах,
часто уплотнены)
легкое снижение интеллекта
гинекомастия
евнуховидное
телосложение
(узкие плечи, широкий таз)
женский тип оволосения лобка,
плохой рост бороды, отсутствие
залысин на лбу
вспыльчивость, импульсивность
Частота: 1 мальчик на
1000 новорожденных
43.
Синдром двойного игрек (47, ХYY )Характерные признаки:
- высокий рост (выше 183 см),
- в остальном фенотипических
отличий от здоровых мужчин
не имеют,
- интеллект нарушен, либо на
нижней границе нормы,
- агрессивное поведение:
склонны к поджогам, воровству,
убийству
43
Частота: 1 мальчик на 1000-900 новорожденных
44.
Синдром трисомии по Х - хромосоме(47, ХХХ)
-физическое развитие не
отличается женской популяции,
- легкое снижение интеллекта,
- нормально беременеют и
рожают здоровых детей
Частота: 1 случай на 1000 новорожденных
44
45. Синдром Шерешевского-Тернера 45,Х0
Клиническая характеристика:•низкий рост,
•снижение интеллекта,
•низкорослость - 98%,
•общая диспластичность (неправильное
телосложение) - 92%,
•бочкообразная грудная клетка - 75%,
•укорочение шеи - 63%,
•низкий рост волос на шее - 57%,
•крыловидные складки кожи в области шеи 46%,
•деформация ушных раковин - 46%,
•аплазия фаланг (укорочены 4 и 5 палец)46%,
•деформация локтевых суставов - 36%,
•широко расставлены соски,
•резкий половой инфантилизм
Частота: 1 случай на
2000 новорожденных
46.
Синдром Дауна•47, ХХ + 21
•47, ХY+ 21
Характерные симптомы:
• Снижение интеллекта –
99%
• Плоское лицо – 90%
• Монголоидный разрез глаз
– 80%
• Эпикант – 40%
• Макроглоссия – 50%
• Косоглазие – 60%
• Мелкие зубы – 65%
• Короткие конечности – 70%
• Низкий рост – 80%
• Подражают хорошему
поведению
• Ласковы
Частота: 1 случай на 650 новорожденных
47. Синдромом Дауна (47,ХХ +21; 47,ХУ+21)
ЕвропеоидНегроид
Монголоид
Дети с синдромом Дауна: общие признаки синдрома Дауна
более заметны, чем расовые различия
(Courtesy of Dr.T.M. Schroeder-Kurth.)
47
48. Риск рождения ребенка с синдромом Дауна в зависимости от возраста матери (цифры для живорожденных рассчитаны в каждой
возрастной группе)Возраст матери
Частота на 1000 родов
Риск
Любой возраст
1,5
1/650
30 лет
1,4
1/700
34 года
2,0
1/500
35 лет
2,2
1/450
36 лет
2,5
1/400
37 лет
4,0
1/250
38 лет
5,0
1/200
39 лет
6,5
1/150
40 лет
10,0
1/100
41 год
12,5
1/80
42 года
16,5
1/60
43 года
20
1/50
44 года
25
1/40
48
49.
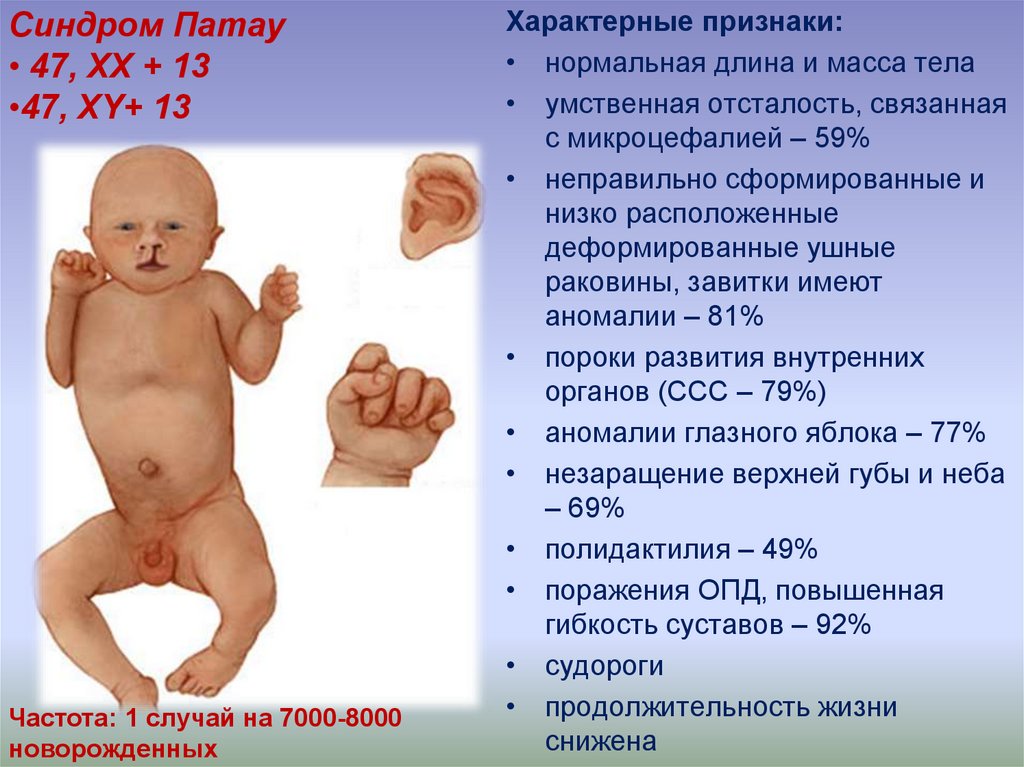
Синдром Патау• 47, ХХ + 13
•47, ХY+ 13
Частота: 1 случай на 7000-8000
новорожденных
Характерные признаки:
• нормальная длина и масса тела
• умственная отсталость, связанная
с микроцефалией – 59%
• неправильно сформированные и
низко расположенные
деформированные ушные
раковины, завитки имеют
аномалии – 81%
• пороки развития внутренних
органов (ССС – 79%)
• аномалии глазного яблока – 77%
• незаращение верхней губы и неба
– 69%
• полидактилия – 49%
• поражения ОПД, повышенная
гибкость суставов – 92%
• судороги
• продолжительность жизни
снижена
50. Основные клинические симптомы трисомии по хромосоме 13 (47, ХY+13) (синдром ПАТАУ)
5051.
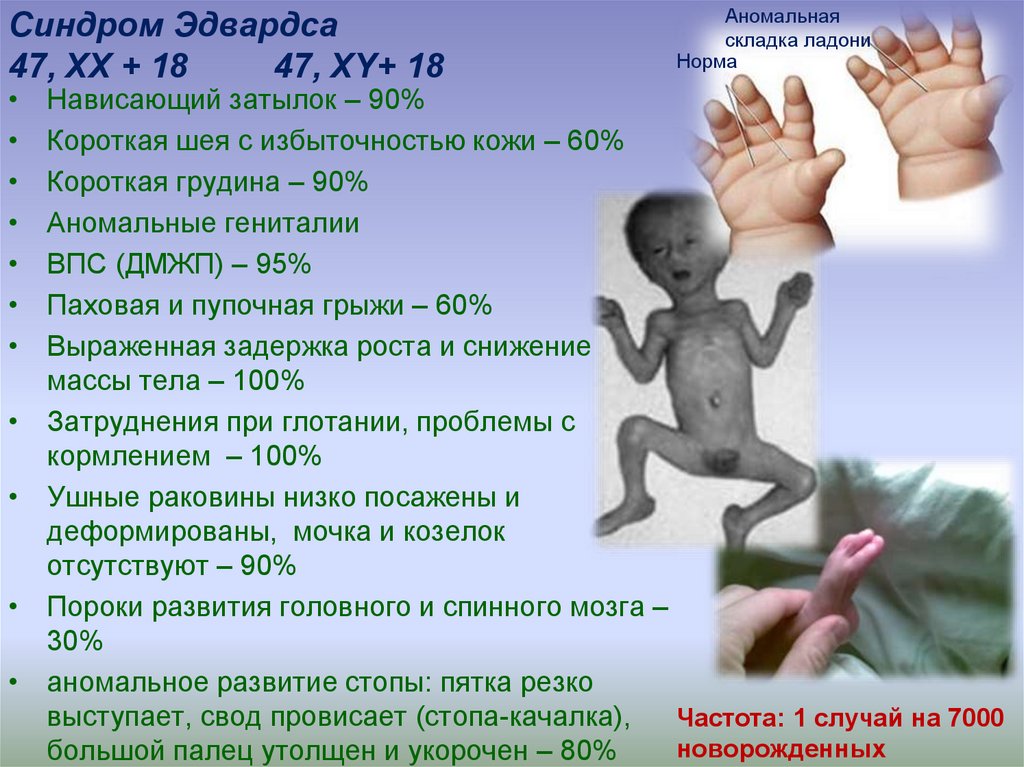
Синдром Эдвардса47, ХХ + 18
47, ХY+ 18
Аномальная
складка ладони
Норма
Нависающий затылок – 90%
Короткая шея с избыточностью кожи – 60%
Короткая грудина – 90%
Аномальные гениталии
ВПС (ДМЖП) – 95%
Паховая и пупочная грыжи – 60%
Выраженная задержка роста и снижение
массы тела – 100%
Затруднения при глотании, проблемы с
кормлением – 100%
Ушные раковины низко посажены и
деформированы, мочка и козелок
отсутствуют – 90%
Пороки развития головного и спинного мозга –
30%
аномальное развитие стопы: пятка резко
выступает, свод провисает (стопа-качалка),
Частота: 1 случай на 7000
новорожденных
большой палец утолщен и укорочен – 80%
52. Основные врожденные пороки при синдроме Эдвардса
двусторонняя лучеваякосорукость с аплазией I
пальца,
укорочение I пальца стоп,
контрактура в тазобедренных
и коленных суставах;
микрогения низко
расположенная и
деформированная ушная
раковина;
флексорное положение
пальцев кисти.
52
53.
ФРАГИЛЬНОСТЬ (ломкость) ПО Х ХРОМОСОМЕвызвана увеличением «нормального» количества дупликаций тринуклеотида ЦГГ
на участке 28 длинного плеча Х хромосомы. Имеются сообщения о развитии
премутации при наличии более 55 повторов (51-54 повтора—пограничное число),
а полная мутация отмечается, когда количество повторов превышает 200.
46, fraX
Частота: 1 случай на 2000-5000
новорожденных. У мальчиков –
53
в 2-3 раза чаще
54. Клеточный=хромосомный= =генетический мозаицизм
явление, когда в соматических клетках одногоорганизма имеется разный набор хромосом
(46,ХХ/47,ХХ+21; 46,ХУ/45,Х0)
Возникает в результате нерасхождения
хромосом во время митоза.
По наследству
мозаицизм не
передается, т.к.
изменения
хромосомного набора
имеют место только в
соматических клетках.
54
55.
Спорадическая мутацияпроисходит во время
ранних делений
эмбриона, которые, таким
образом, формируются из
смеси генетически
различных клеток.
В зависимости от распределения мутировавших клеток между
трофобластом и эмбриобластом различные органы будут
поражены мутацией с разной степенью тяжести.
55
56. Структурные аномалии хромосом:
• Изохромосомы 46,iX = 46,изоХ• Делеции
46,ХХ,5р- = 46,ХУ,del 5р
• Транслокации 46,ХХ,t (9:22)
• Инверсии
46,ХХ,inv16
• Избыток
46,ХХ,dup18q
• Нехватки
46,ХХ,del 5p = 46, ХХ,5р• Кольцевые хромосомы 46,ХХ,r 18
46,ХУ,r 18
56
57. Синдромом частичной трисомии проксимального отдела длинного плеча 14 хромосомы
•черепно-лицевые аномалии;•умственная отсталость;
•пороки сердца;
•дупликация дистальной части 14q
57
58. Фенотипические проявления при делециях
Фенотипические признакиТип делеции
4p-
5p-
18 p-
18 g-
«Кошачий крик»
-
+
-
-
Слабоумие
+
+
+
+
Пороки развития мозга
+
-
-
-
Микроцефалия
+
(+)
-
+
Колобома радужной оболочки
+
-
-
-
Гипертелоризм
+
+
(+)
+
Эпикант
(+)
(+)
(+)
-
Птоз
+
-
(+)
-
Косоглазие
(+)
(+)
(+)
-
Гипоплазия носа и верхней челюсти
-
-
-
(+)
Аномалия ушных раковин
+
(+)
(+)
(+)
Атрезия наружного слухового прохода
-
-
-
(+)
Микрогнатия
+
(+)
+
-
Расщепление неба
+
-
-
-
Порок сердца
(+)
-
-
(+)
Гипоспадия
+
-
-
-
Кариес
-
-
+
-
Гипоплазия папиллярных линий
+
-
-
- 58
Увеличение числа завитков на пальцевых подушечках
-
(+)
-
+
59.
Чем больше генетического материала тем хуже. Впервую очередь страдает интеллект.
В результате хромосомных мутаций и аберраций
возникает дисбаланс генетического материала,
который приводит к нарушению психического и
физического развития.
Аномалии по крупным хромосомам встречаются
реже, чем по мелким. Нехватки генетического
материала переносятся тяжелее. Частота
встречаемости хромосомных нарушений зависит
от того к какому хроматину относится нарушение.
Наиболее часто встречаются нарушения по
8,13,18,21,Х хромосомам.В этих хромосомах
больше гетерохроматина.
59
60. Евгеника (греч.eugenes=хорошего рода, знатного происхождения, хорошей расы) - учение о наследственном здоровье человека и путях
Евгеника (греч.eugenes=хорошего рода,знатного происхождения, хорошей расы) учение о наследственном здоровье человека и
путях его улучшения
60
61. НОМЕНКЛАТУРА УМСТВЕННОЙ ОТСТАЛОСТИ ( умственно отсталый человек это «индивид, неспособный к независимой социальной адаптации»)
Степень дефектаАмериканские
названия
Умственный возраст
в годах (взрослые)
Слабая
Дебил
7-10
Тяжелая
(среднеранговый)
Слабоумный
3-6
Тяжелая
(низкоранговый)
Идиот
0-2
61
62.
• Более высокая частота среди умственноотсталых приходится на долю мальчиков (1,8
на 1000), объясняется тем, что этот признак
сцеплен
с
Х-хромосомой.
46,fraХ
(фрагильность=ломкость Х хромосомы).
• Болезни связанные с нарушением репарации
ДНК:-многие
синдромы
с
аутосомной
нестабильностью
или
ломкостью.
При
умственной
отсталости
обнаружили
нестабильность аутосом:2,6,7,13,16.
62