












 medicine
medicineSimilar presentations:










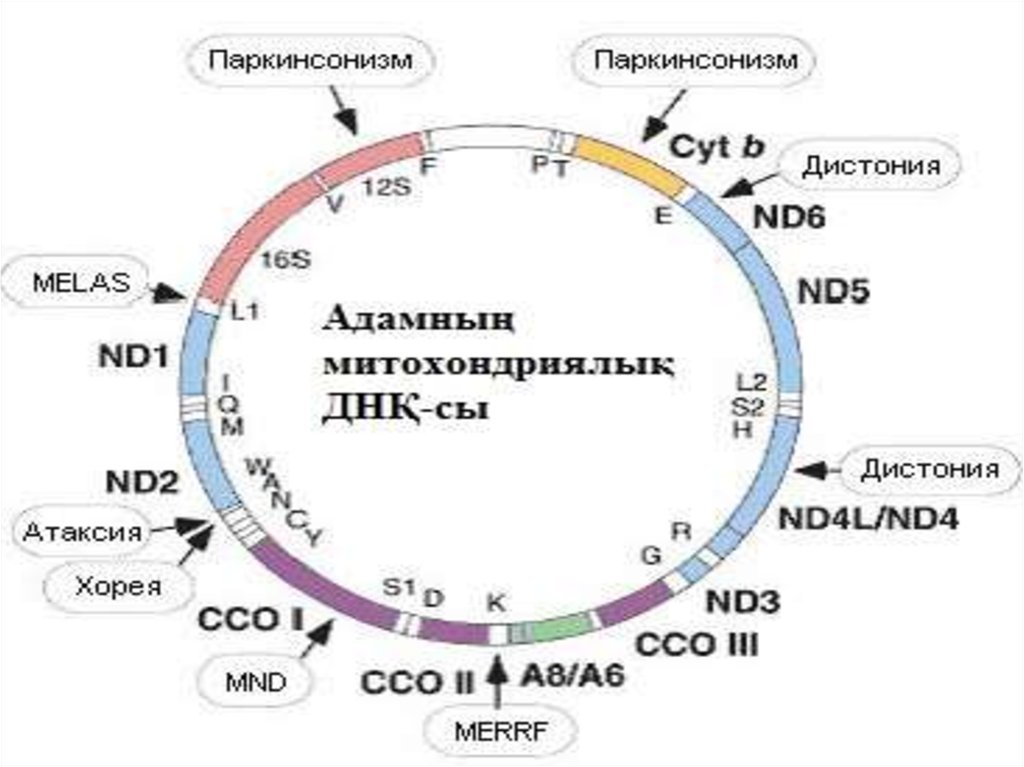
Митохондриялыќ аурулар
1. С¤Ж Таќырыбы: «Митохондриялыќ аурулар»
“Астана медициналық университеті ”АҚМолекулалық биология және медициналық генетика кафедрасы
Орындаған: 104- ЖМ топ студенті
Бекболат Галымбек
Оқытушы: А.Н. Закирияевна
2017 жыл
2. Жоспар
Кіріспе. Тарихи анықтама.Митохондрияныңқызметі мен құрылысы.
2. Митохондриялық аурулардың жіктелуі.
3. Митохондриялық аурулар патогенезі.
4. Митохондриялық аурулар клиникасы.
5. Митохондриялық аурулар диагностикасы.
6. Митохондриялық ауруларды емдеу жолдары.
7. Кейбір синдромдарға сипаттама.
8. Қорытынды
Пайдаланылған әдебиеттер.
1.
3. Кіріспе. Митохондрияныњ ќызметі мен ќ±рылысы. Тарихи аныќтама.
Митохондриялық аурулар – митохондридің қызметінің дефектісінебайланысты болатын,жасушадағы энергетикалық қызметтің
бұзылуына әкелетін тұқым қуалайтын аурулардың тобы
4.
Митохондрияның негізгі қызметтері:•АТФ түрінде жасушаларға энергия өндіру;
•Май қышқылдарының β тотығуы;
•Үш карбонқышқылының циклі;
•Жасуша ішілік сигнал беруге,апоптозға қатысу;
•Аминқышқылдар,липидтер,стероидтар,нулеотидтер метаболизміне қатысу;
5.
6.
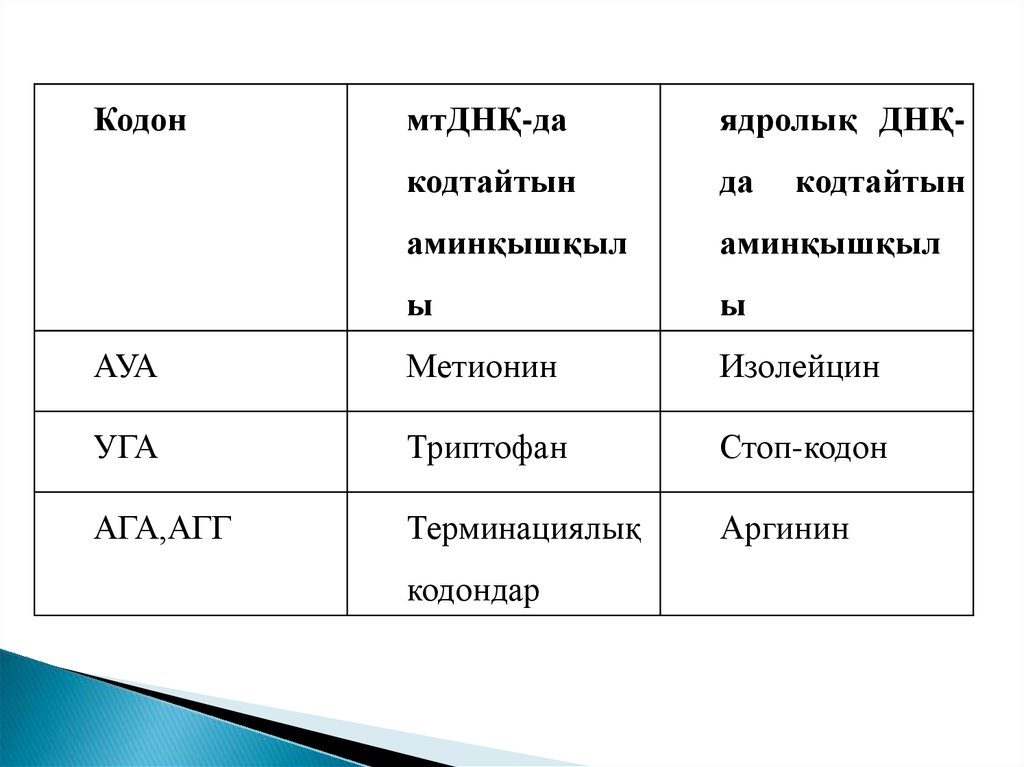
КодонмтДНҚ-да
ядролық ДНҚ-
кодтайтын
да
аминқышқыл
аминқышқыл
ы
ы
АУА
Метионин
Изолейцин
УГА
Триптофан
Стоп-кодон
АГА,АГГ
Терминациялық
Аргинин
кодондар
кодтайтын
7.
Алғашқы митохондриялық аурулар мтДНҚашылмай тұрып белгілі болған.
1958 – синдром Кернс-Сейра
1962 - Люфт ауруы
1963-мтДНҚ-ның ашылуы
1981 – адамның митохондриялық геномы оқылды
1988 – мтДНҚ-дағы мутациялар белгілі бола
бастады
8. Митохондриялыќ аурулардыњ жіктелуі.
Митохондриялық ауруларМитохондриялық ДНҚ-ның
мутациясы салдарынан
туындайтын аурулар
Митохондриялық ДНҚ –ның
өзгерісіне әкелетін ядролық
ДНҚ бұзылыстарына
байланысты туындайтын
аурулар
ядролық ДНҚ-ның
дефектісіне байланысты
туындайтын аурулар
9.
Митохондриялық ДНҚ-да мутаци ядролықДНҚ-ға қарағанда 5 есе жиі болады
Митохондрий өзіне жұтылған оттегінің 90%
сіңіреді.
Митохондрий айналасында мембрананы
бұзушы бос радикалдар түзіледі.
Митохондрий геномы гистондық ақуыздармен
қорғалмаған;
Репарация митохондрийде ядроға қарағанда
жетілмеген
10.
Примеры некоторых синдромовСиндром Лебера: LHON (1871 г.)
- наследуемая по материнской линии потеря зрения
происходит у людей 20-30 лет вследствие
• атрофии зрительного нерва и
• дегенерации ганглиозного слоя клеток ретины
Заболевание связано с передаваемой от матери мутацией
митохондриальной ДНК в одном из ND генов (комплекс I).
В 70% случаев это G11778A (ND4), а в Японии в 90%
в 13% случаев G3460A (ND1);
в 14% случаев T14484C (ND6)
Мутация находится в гомоплазматическом состоянии
11.
ДНК-диагностика синдрома Лебера в семье Nпроведена нами впервые в 2006 году
745 п.н.
634 п.н.
здоровый сестра мать
человек пробанда
G11778
G11778A замена
пробанд с
синдромом
Лебера
12.
Загадки синдрома Лебера:??
В 80-85% случаев поражаются мужчины
??
Лишь у 50% мужчин и 10% женщин носителей
патогенных мутаций комплекса I в
действительности происходит потеря зрения
??
Чаще всего мутации, ведущие к синдрому
Лебера, встречаются в мтДНК гаплогруппы J;
эту группу несут около 15% европейцев
(Х хромосома несет какой-то локус чувствительности ?)
В формировании заболевания участвуют
какие-то дополнительные факторы ( ???)
13.
Мутации генов транспортной РНКСамая часто встречающаяся точечная мутация:
А3243G в лейциновой тРНК
Обнаружена у большинства больных с синдромом
Миопатия
энцефалопатия
MELAS
лактатацидоз
инсультоподобные
(stroke-like)
эпизоды
Мутация встречается исключительно в
гетероплазматическом состоянии
???
В одних семьях А3243G вызывает преимущественно
кардиомиопатию, в других – диабет и глухоту, в
третьих PEO, в четвертых - энцефалопатию
14.
Впервые ДНК-диагностика синдромаMELAS была проведена нами в 2007 году
I брак
1ый ребенок
1988-2000
Кардиопатия,
ЗПР, ЗФР.
Умерла
скоропостижно
после травмы
Мама: фенотипически здоровая
женщина очень маленького роста
II брак
2ой ребенок
3ий ребенок
1991-2007
родился в 1998
Менингоэнцефалит Прогрессирующая
миопатия,
Умер от
ишемического
миокардиоинфаркта обоих
дистрофия
полушарий
мозжечка
Митохондриопатия??
Обнаружена мутация MELAS у сына (80% мутантных
молекул в крови) у мамы (40% )
15.
Синдром Лея – тяжелейшеенейродегенеративное заболевание:
- симметричные некротические повреждения в
субкортикальных областях ЦНС – базальных
ганглиях, таламусе, стволе мозга, спинном мозге;
- демиелинизация, сосудистая пролиферация и
«глиозис»;
- моторная и умственная регрессия,
атаксия, дистония, аномальное дыхание
Заболевание начинается в раннем
детстве, редко во взрослом состоянии;
Смерть наступает обычно через два года после
начала заболевания