































































 biology
biologySimilar presentations:
")







")

Наследственная изменчивость. Понятие о наследственных патологиях у человека и их профилактике. Лекция №14
1.
ГБПОУ ЛО «Центр непрерывного профессионального медицинскогоразвития Ленинградской области»
Лекция №14
Наследственная изменчивость.
Понятие о наследственных патологиях
у человека и их профилактике
Преподаватель: Видерникова Е.А.
2.
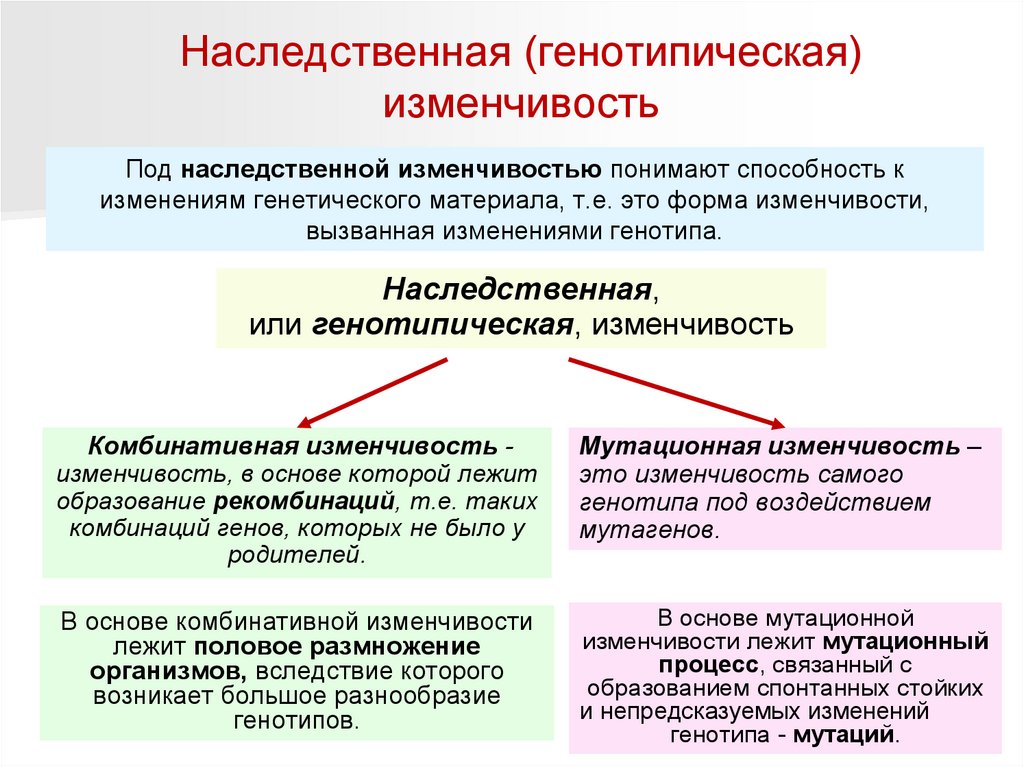
Наследственная (генотипическая)изменчивость
Под наследственной изменчивостью понимают способность к
изменениям генетического материала, т.е. это форма изменчивости,
вызванная изменениями генотипа.
Наследственная,
или генотипическая, изменчивость
Комбинативная изменчивость изменчивость, в основе которой лежит
образование рекомбинаций, т.е. таких
комбинаций генов, которых не было у
родителей.
Мутационная изменчивость –
это изменчивость самого
генотипа под воздействием
мутагенов.
В основе комбинативной изменчивости
лежит половое размножение
организмов, вследствие которого
возникает большое разнообразие
генотипов.
В основе мутационной
изменчивости лежит мутационный
процесс, связанный с
образованием спонтанных стойких
и непредсказуемых изменений
генотипа - мутаций.
3.
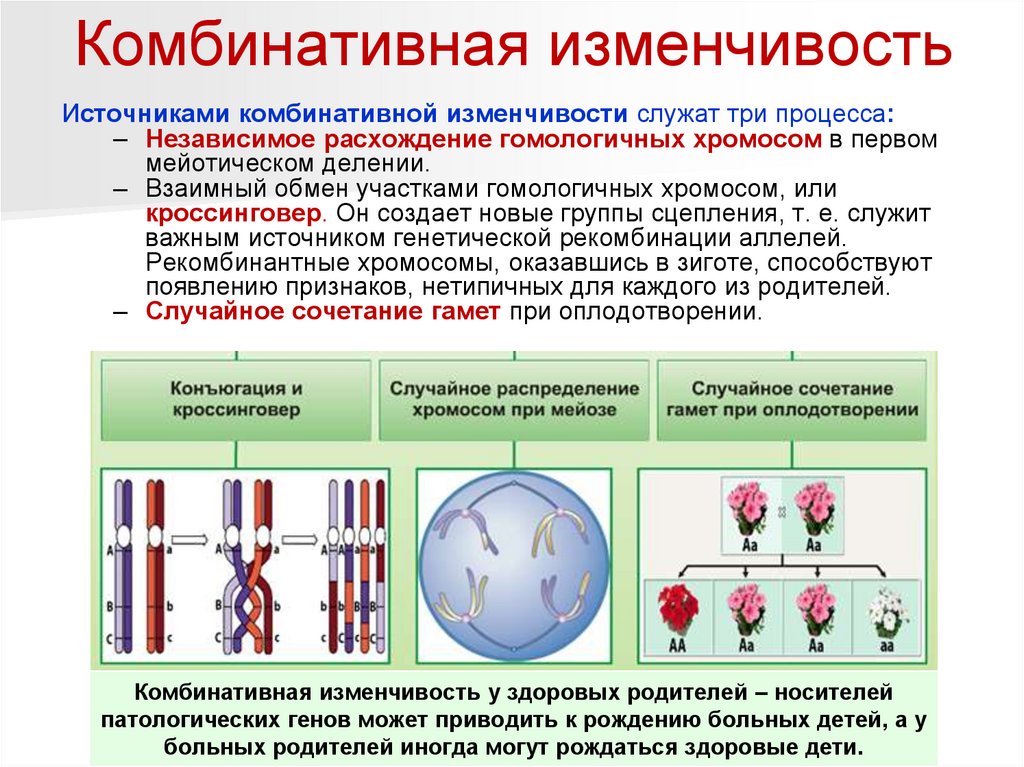
Комбинативная изменчивостьИсточниками комбинативной изменчивости служат три процесса:
– Независимое расхождение гомологичных хромосом в первом
мейотическом делении.
– Взаимный обмен участками гомологичных хромосом, или
кроссинговер. Он создает новые группы сцепления, т. е. служит
важным источником генетической рекомбинации аллелей.
Рекомбинантные хромосомы, оказавшись в зиготе, способствуют
появлению признаков, нетипичных для каждого из родителей.
– Случайное сочетание гамет при оплодотворении.
Комбинативная изменчивость у здоровых родителей – носителей
патологических генов может приводить к рождению больных детей, а у
больных родителей иногда могут рождаться здоровые дети.
4.
Комбинативная изменчивостьКомбинативная изменчивость - важнейший источник
большого наследственного разнообразия,
характерного для живых организмов.
Однако перечисленные источники изменчивости не
порождают существенных для выживания
стабильных изменений в генотипе, которые
необходимы, согласно эволюционной теории, для
возникновения новых видов.
Такие изменения возникают в
результате мутаций.
5.
Мутационная изменчивостьОсновным источником многообразия наследственных признаков и
их непрекращающейся эволюции служит мутационная
изменчивость.
Мутации (лат. mutatio - перемена) - это внезапно возникающие
стойкие изменения генетического материала, приводящие к
изменению тех или иных наследственных признаков организма.
Модификации:
Зависят от силы фактора;
Адекватны вызвавшему их
фактору;
Не наследуются;
Исчезают после
прекращения действия
фактора.
Мутации:
Случайны и не зависят от
силы фактора;
Не адекватны вызвавшему
их фактору;
Наследуются;
Сохраняются после
прекращения действия
фактора.
Отличия мутаций от модификаций
6.
Мутационная теория Хуго Де ФризаОсновные положения мутационной теории разработаны Хуго де Фризом в
1901-1903 гг. (он же предложил термин “мутация”):
1.
Мутации возникают внезапно,
скачкообразно, как дискретные изменения
признаков.
2.
В отличие от ненаследственных изменений
мутации представляют собой качественные
изменения, которые передаются из
поколения в поколение.
3.
Мутации проявляются по-разному и могут
быть как полезными, так и вредными, как
доминантными, так и рецессивными.
4.
Вероятность обнаружения мутаций
зависит от числа исследованных особей.
5.
Сходные мутации могут возникать
повторно.
Хуго де Фриз
(1848-1935)
6. Мутации ненаправленны (спонтанны), т.е. мутировать может любой
участок хромосомы, вызывая изменения как незначительных, так и жизненно
важных признаков.
7.
Мутационная изменчивостьСуществует много мутаций и у человека. Именно мутациями
обусловлен полиморфизм человеческих популяций:
– различная пигментация кожи, волос, окраска глаз, разрез глаз
– форма носа, ушей, подбородка
– группы крови и т. д.
В результате мутаций появляются и наследственные аномалии в
строении тела, и наследственные болезни человека.
Медицинская генетика вплотную занимается этой проблемой!!!
Наследственная патология - часть наследственной изменчивости,
накопившейся за время эволюции человека.
Человек, став биологическим видом Homo sapiens, как бы заплатил за
“сапиентацию” своего вида накоплением патологических мутаций.
На основе этих положений формулируется одна из главных концепций
медицинской генетики об эволюционном накоплении патологических
мутаций в человеческих популяциях
8.
Мутагенные факторыМутагены (мутагенные факторы) – это факторы,
способные вызывать мутации.
экзомутагены
(химические,
биологические,
физические);
эндомутагены – нарушения
репликации, транскрипции,
трансляции, репарации, генымутаторы.
Мутагенные факторы по происхождению бывают:
– Физическими;
– Химическими;
– Биологическими.
9.
Физические мутагеныК физическим мутагенам относятся различные
виды излучений, температура, влажность и др.
Основные механизмы действия:
– нарушение структуры генов и хромосом;
– образование свободных радикалов, вступающих в химическое взаимодействие
с ДНК;
– разрывы нитей ахроматинового веретена деления;
– образование димеров нуклеотидов.
Наибольшее значение имеют
ионизирующие излучения, в частности
рентгеновское – радиационный
мутагенез .
В настоящее время установлено, что
наследственные изменения
обусловливаются всеми видами
проникающей радиации.
10.
Химические мутагеныХимические
физических.
мутагены
были
открыты
позже
Приоритет открытия химических мутагенов принадлежит
советским исследователям:
- В 1933 г. В.В. Сахаров получил мутации путем
действия йода, в 1934 г. М. Е. Лобашев - применяя
аммоний.
- В 1946 г. советский генетик И.А. Рапопорт
обнаружил сильное мутагенное действие формалина и
этиленимина, а английская исследовательница
Ш. Ауэрбах - иприта.
11.
Химические мутагеныХимические мутагены способны вызывать
мутации всех типов (преимущественно генные).
Например, алкалоид колхицин разрушает
веретено деления, что приводит к удвоению
количества хромосом в клетке.
Газ иприт, используемый как химическое
оружие, повышает частоту мутаций у
экспериментальных мышей до 90 раз.
Известно множество химических мутагенов:
- природные органические и неорганические
вещества (нитриты, нитраты, алкалоиды,
гормоны, ферменты и др.)
- продукты промышленной переработки
природных соединений – угля, нефти;
- синтетические вещества – инсектициды,
пестициды, пищевые консерванты,
лекарственные вещества;
- некоторые метаболиты организма человека
– кортикостероидные, половые гормоны.
12.
13.
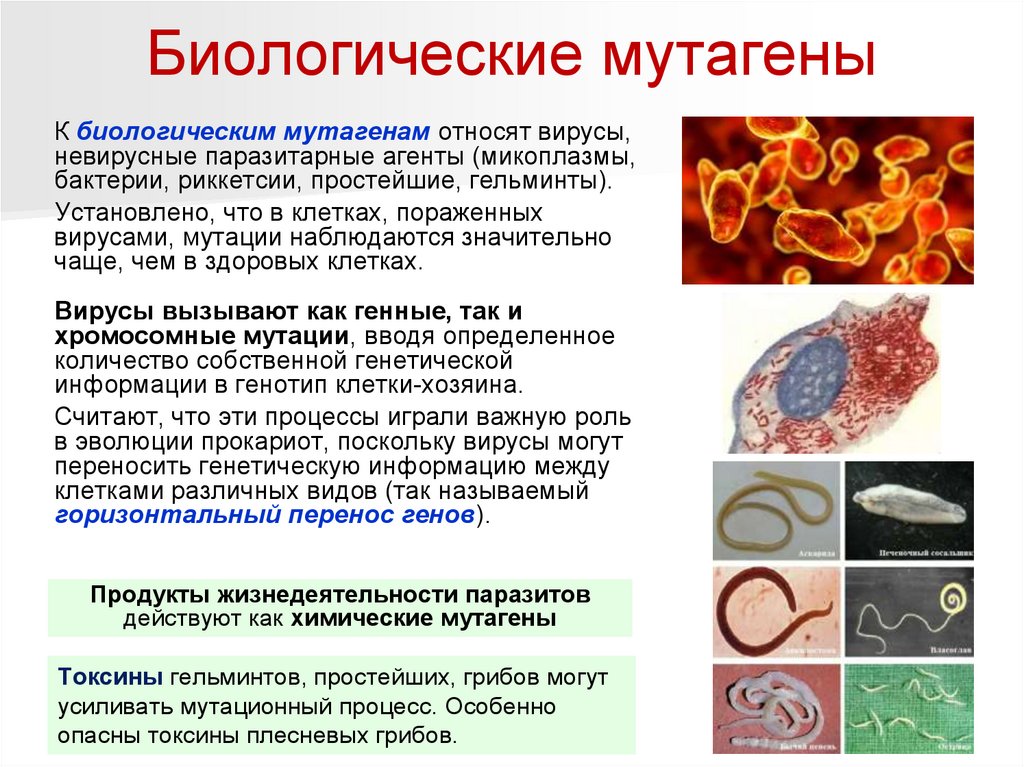
Биологические мутагеныК биологическим мутагенам относят вирусы,
невирусные паразитарные агенты (микоплазмы,
бактерии, риккетсии, простейшие, гельминты).
Установлено, что в клетках, пораженных
вирусами, мутации наблюдаются значительно
чаще, чем в здоровых клетках.
Вирусы вызывают как генные, так и
хромосомные мутации, вводя определенное
количество собственной генетической
информации в генотип клетки-хозяина.
Считают, что эти процессы играли важную роль
в эволюции прокариот, поскольку вирусы могут
переносить генетическую информацию между
клетками различных видов (так называемый
горизонтальный перенос генов).
Продукты жизнедеятельности паразитов
действуют как химические мутагены
Токсины гельминтов, простейших, грибов могут
усиливать мутационный процесс. Особенно
опасны токсины плесневых грибов.
14.
Биологические мутагеныВирусы ветряной оспы, гриппа,
герпеса, паротита, саркомы Рауса –
вызывают генные мутации ,
хромосомные аберрации, а также
поражают репаративные системы.
Непатогенные вирусы играют
существенную роль в генетических
рекомбинациях.
Вакцины могут
вызвать генные и
хромосомные мутации
после вакцинации.
Последний раз ретровирус изменил
геном человека более 100 тысяч лет
назад. Однако за время эволюции это
происходило много раз, поэтому ДНК
человека на целых 8% состоит из
кусочков ДНК древних ретровирусов,
которые заражали половые клетки
наших предков.
единственная возможность для изменения
человеческой ДНК – это использование в составе
вакцин фермента интегразы, способного объединить
две молекулы ДНК в одну. Но производители не
используют этот фермент при изготовлении
вакцин. Такой способностью обладают только
ретровирусы
15.
КомутагеныКомутагены – токсические вещества, которые сами не вызывают
мутации, но существенно изменяют влияние мутагенных факторов
физической, химической и биологической природы.
Комутагенами являются
вещества, используемые в
пищевых добавках, косметике,
СМС, лекарственных
препаратах.
Токсические вещества
приводят к модификационной
изменчивости и изменяют
норму реакции.
Вместе с мутагенами они
способствуют увеличению
генетического груза человека.
Е110 или «Солнечный закат»
используется для придания продуктам
желтого и оранжевого оттенков.
Натриевая соль (начальное вещество
Судан 1), имеющая вид оранжево-красного
порошка или гранул, хорошо растворимых
в водной среде. Вещество не имеет
запаха и характерного вкуса.
16.
АнтимутагеныАнтимутагены – биологически активные вещества, введение которых в
клетку препятствует действию мутагенов, т.е. это вещества группы
хемопревенторов, способные увеличивать устойчивость человека к
разнообразным воздействиям, в том числе и мутагенным.
Основные пути действия
антимутагенов:
1) нейтрализация мутагена до его
взаимодействия с ДНК;
2) предотвращение образования в
процессе метаболической активности
мутагенов из нетоксичных
предшественников;
Делают мутагены
неактивными
3) активация защитных ферментных
систем организма;
Изменяют влияние
мутагенов до неопасного
4) предотвращение ошибок в процессе
репликации ДНК;
5) активация световой и темновой
репарации ДНК.
Усиливают систему
исправления повреждений
в организме
17.
АнтимутагенезЕстественный антимутагенез:
1) четное количество хромосом в
диплоидному наборе (в гетерозиготы
рецессивные гены не проявляются).
2) повторы некоторых генов,
кодирующих рРНК тРНК, гистоновые
белки хроматина;
3) триплетность генетического кода.
4) вырожденность генетического кода замена второго нуклеотида в кодоне
вызывает 100% мутаций, замена
третьего нуклеотида в кодоне вызывает
36% мутаций.
5) репарация первичной структуры ДНК.
Искусственный антимутагенез
- применение веществ и физических
факторов, которые снижают частоту
мутаций:
1)
2)
3)
4)
5)
витаминов (С, Е, А, Р, В9)
аминокислот (метионина,
глутаминовой кислоты)
серотонина, резерпина;
растительных препаратов
(китайского лимонника, жень-шеня,
родиолы);
физических факторов (дневной
свет).
18.
Классификация мутацийМутации классифицируют:
– по причинам, их вызвавшим;
– по характеру мутировавших клеток;
– по исходу для организма;
– по характеру изменений генетического
материала.
19.
Классификация мутаций1. Соматические и генеративные
мутации
По характеру мутировавших клеток мутации подразделяют на
соматическое и генеративные.
Биологическое значение их неравноценно и связано с характером
размножения организмов.
Соматические мутации происходят в
соматических клетках и проявляются
у самой особи.
При делении мутировавшей
соматической клетки новые свойства
передаются ее потомкам - клону.
При половом размножении признаки,
появившиеся в результате
соматических мутаций, потомкам не
передаются и в процессе эволюции
никакой роли не играют.
Генеративные мутации происходят
в клетках, из которых развиваются
гаметы, или в половых клетках.
Новый признак проявится в
ближайшем или последующих
поколениях.
Генеративные мутации
являются материалом для
естественного отбора.
20.
Соматические мутацииОднако в индивидуальном развитии соматические мутации могут влиять на
формирование признака: чем в более ранней стадии развития возникнет
соматическая мутация, тем больше участок ткани, несущий данную
мутацию.
Такие особи называются мозаиками.
Например, мозаиками являются люди, у
которых цвет одного глаза отличается
от цвета другого, или животные
определенной масти, у которых на теле
появляются пятна другого цвета, и т. п.
Соматические мутации могут быть
причиной:
– доброкачественных и злокачественных
новообразований;
– лейкозов;
– некоторых болезней тканей и органов,
где клеточный пул быстро обновляется
(пароксизмальная ночная
гемоглобинурия, макроглобулинемия
Вальденстрема и др.болезни крови и
иммунной системы).
Не исключено, что соматические
мутации, влияющие на метаболизм,
являются одной из причин старения.
21.
Генеративные мутацииГенеративные мутации происходят в клетках, из которых
развиваются гаметы, или в половых клетках.
Мутации передаются по наследству
при половом размножении и
выявляются фенотипически у
потомков.
Новый признак проявится в
ближайшем или последующих
поколениях.
Генеративные мутации являются
материалом для естественного
отбора.
22.
Классификация мутаций по исходудля организма
2. По исходу для организма все
мутации подразделяют на:
– Отрицательные:
летальные (несовместимые с
жизнью);
полулетальные (снижающие
жизнеспособность организма);
– Нейтральные;
– Положительные (повышающие
приспособленность и жизнестойкость
организма). Встречаются
относительно редко, однако именно
они являются элементарным
материалом, лежащим в основе
прогрессивной эволюции.
23.
Классификация мутаций3. По причинам мутации подразделяются на:
Спонтанные мутации возникают
под влиянием неизвестных
природных факторов, чаще всего как
результат ошибок при
воспроизведении генетического
материала (ДНК или РНК).
Причинами спонтанных мутаций
могут быть естественный
радиационный фон, космические
лучи, достигающие поверхности
Земли, и другие причины.
Частота спонтанного мутирования у
каждого вида генетически
обусловлена и поддерживается на
определенном уровне.
Спонтанные мутации происходят
относительно редко и служат
исходным материалом для
естественного отбора.
Индуцированные мутации
возникают при направленном
воздействии на организм
мутагенными факторами.
Впервые индуцированные
мутации были получены Г. А.
Надсоном и Г. С. Филипповым
(1925) при облучении грибов
радием и Г. Меллером (1927)
при облучении мух дрозофил
рентгеновскими лучами.
Индуцированные мутации
служат исходным материалом
для искусственного отбора.
24.
Классификация мутаций по характеруизменения генетического материала
4. По характеру изменения генетического аппарата
различают мутации:
– геномные (изменение числа хромосом);
– хромосомные (изменение структуры хромосом,
хромосомные аберрации);
– генные или точковые (изменение молекулярной
структуры гена).
Отдельно выделяют цитоплазматические мутации,
причиной которых является изменчивость определенных
органоидов цитоплазмы (митохондрий, плазмид, пластид),
содержащих ДНК или РНК
25.
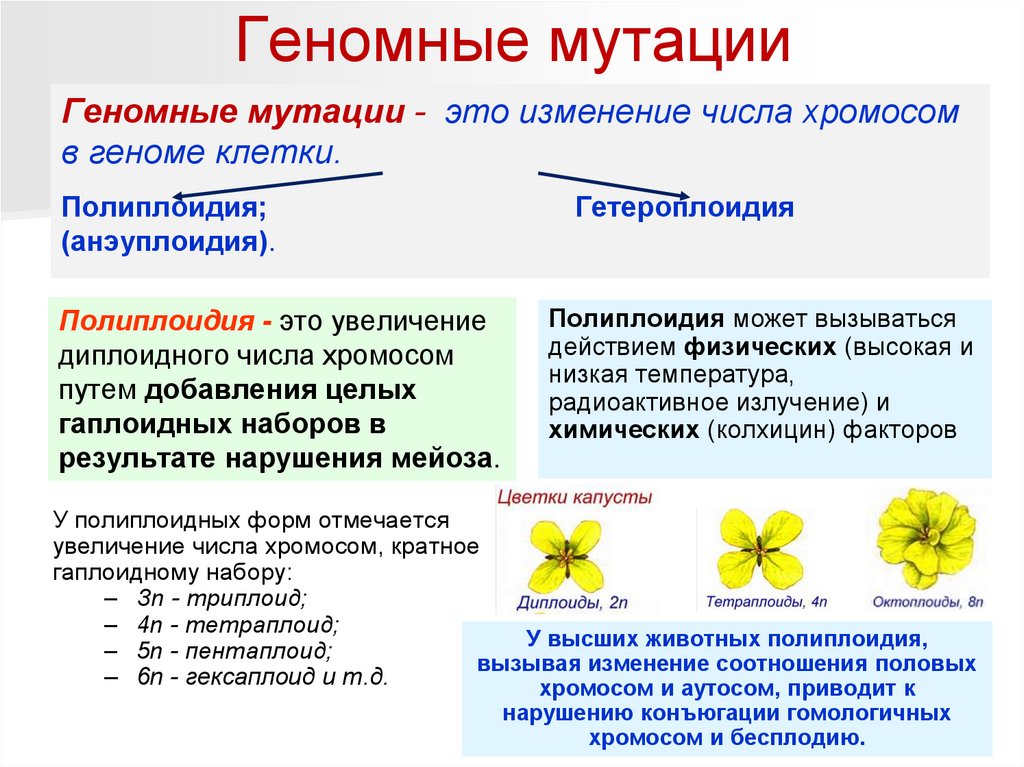
Геномные мутацииГеномные мутации - это изменение числа хромосом
в геноме клетки.
Полиплоидия;
(анэуплоидия).
Полиплоидия - это увеличение
диплоидного числа хромосом
путем добавления целых
гаплоидных наборов в
результате нарушения мейоза.
Гетероплоидия
Полиплоидия может вызываться
действием физических (высокая и
низкая температура,
радиоактивное излучение) и
химических (колхицин) факторов
У полиплоидных форм отмечается
увеличение числа хромосом, кратное
гаплоидному набору:
– Зn - триплоид;
– 4n - тетраплоид;
У высших животных полиплоидия,
– 5n - пентаплоид;
вызывая изменение соотношения половых
– 6n - гексаплоид и т.д.
хромосом и аутосом, приводит к
нарушению конъюгации гомологичных
хромосом и бесплодию.
26.
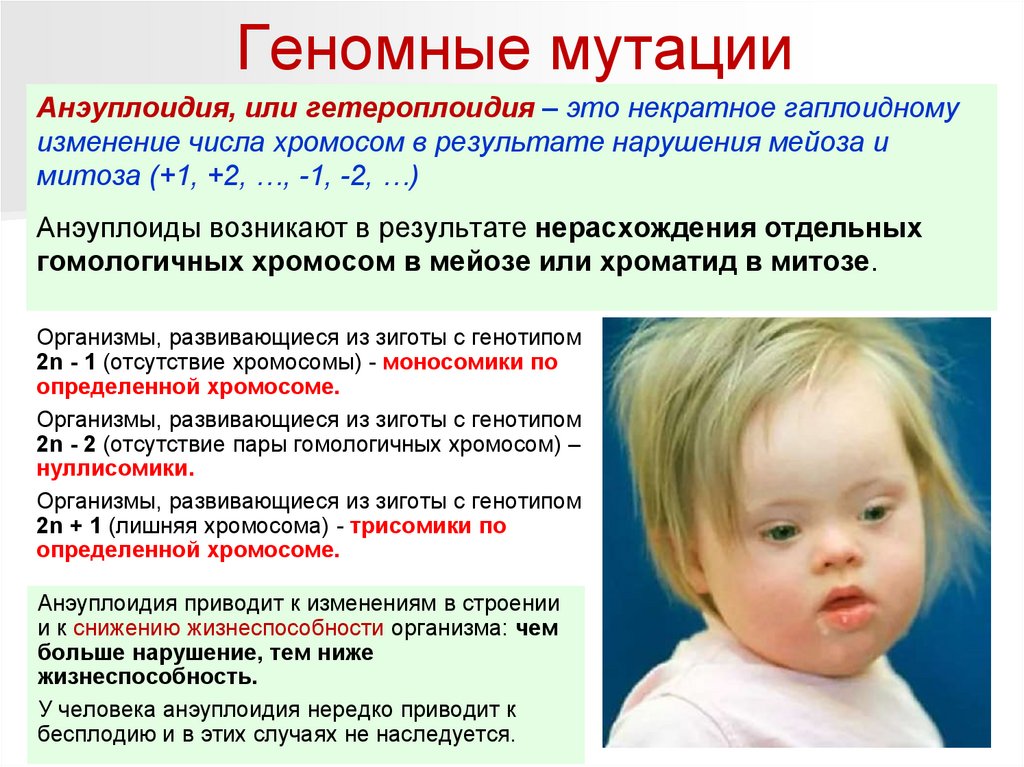
Геномные мутацииАнэуплоидия, или гетероплоидия – это некратное гаплоидному
изменение числа хромосом в результате нарушения мейоза и
митоза (+1, +2, …, -1, -2, …)
Анэуплоиды возникают в результате нерасхождения отдельных
гомологичных хромосом в мейозе или хроматид в митозе.
Организмы, развивающиеся из зиготы с генотипом
2n - 1 (отсутствие хромосомы) - моносомики по
определенной хромосоме.
Организмы, развивающиеся из зиготы с генотипом
2n - 2 (отсутствие пары гомологичных хромосом) –
нуллисомики.
Организмы, развивающиеся из зиготы с генотипом
2n + 1 (лишняя хромосома) - трисомики по
определенной хромосоме.
Анэуплоидия приводит к изменениям в строении
и к снижению жизнеспособности организма: чем
больше нарушение, тем ниже
жизнеспособность.
У человека анэуплоидия нередко приводит к
бесплодию и в этих случаях не наследуется.
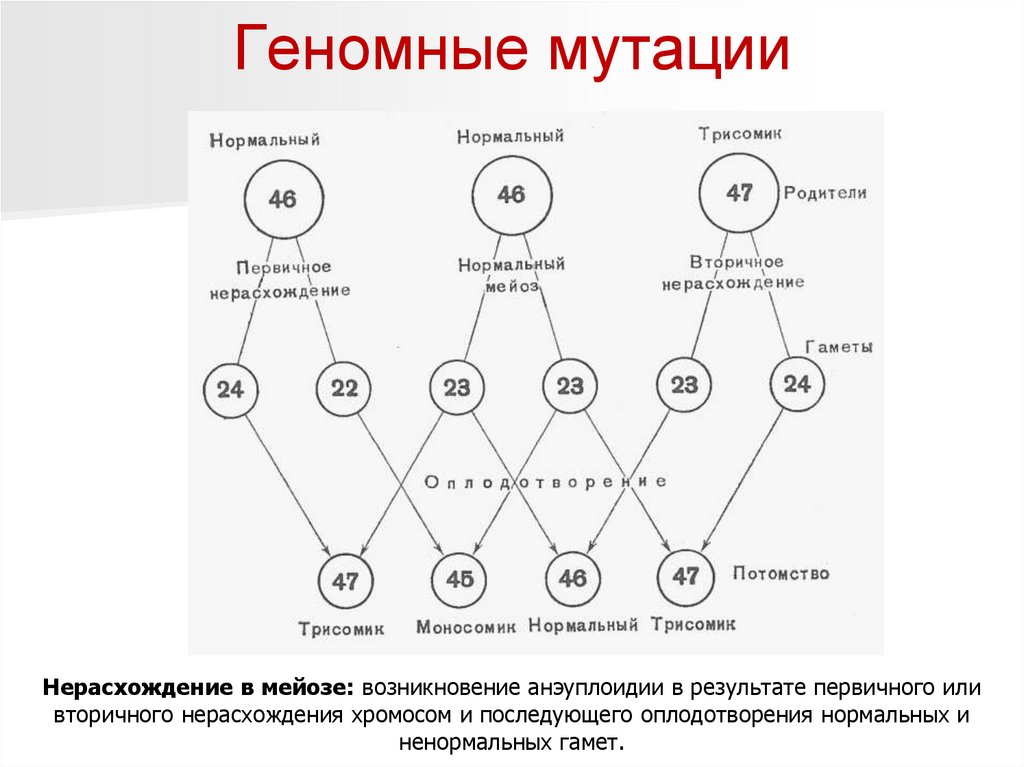
27.
Геномные мутацииНерасхождение в мейозе: возникновение анэуплоидии в результате первичного или
вторичного нерасхождения хромосом и последующего оплодотворения нормальных и
ненормальных гамет.
28.
Хромосомные мутацииХромосомные мутации, или хромосомные
перестройки (аберрации), выражаются в изменении
структуры хромосом.
Хромосомные аберрации связаны с разрывами
хромосом, возникающими в результате повреждения
ДНК (радиацией или химикатами) или в связи с
механизмами рекомбинации.
Клетки обладают системами, которые узнают и при
возможности устраняют разрывы хромосом.
Репарация может происходить соединением двух
разорванных концов или покрытием разорванного
конца теломерой.
29.
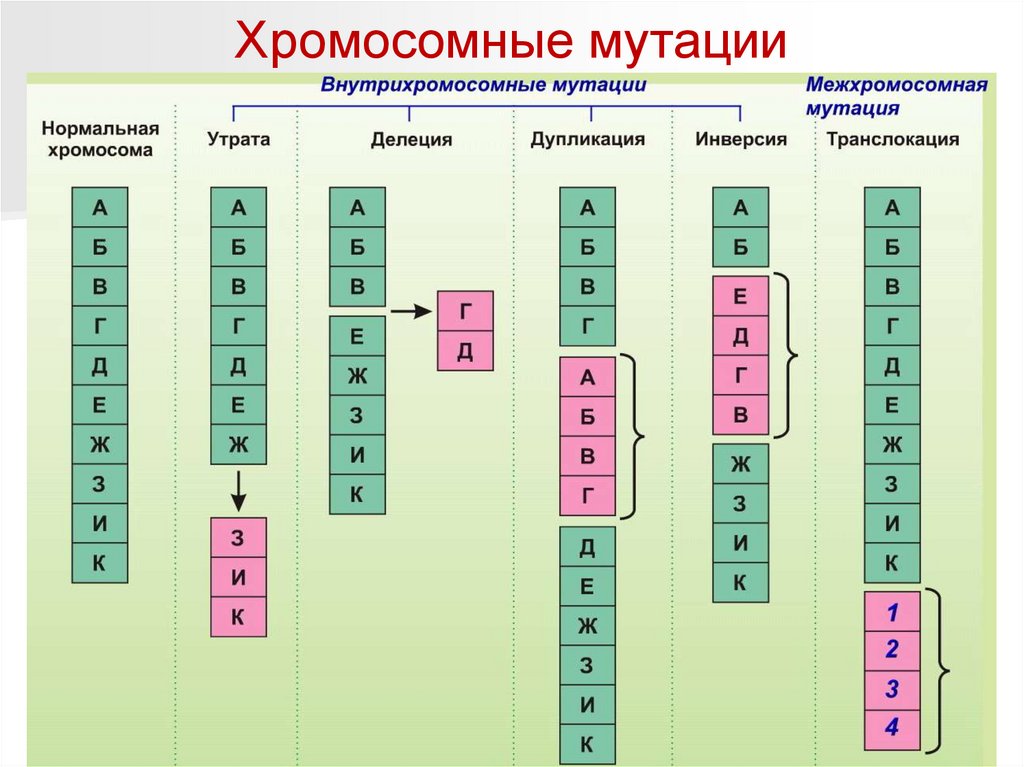
Хромосомные мутации30.
Хромосомные мутацииИзвестны хромосомные аберрации разных типов:
– нехватки — это потеря концевых участков хромосомы;
– делеции (del) — выпадение участка хромосомы в
средней ее части;
– дупликации (dup) — двух- или многократное
повторение набора генов, локализованных в
определенном участке хромосомы;
– инверсии (inv) — поворот участка хромосомы на 180°, в
результате чего в этом участке гены расположены в
последовательности, обратной по сравнению с обычной;
– транслокации (t) — перенос участка к другому концу
той же хромосомы либо к другой, негомологичной
хромосоме.
31.
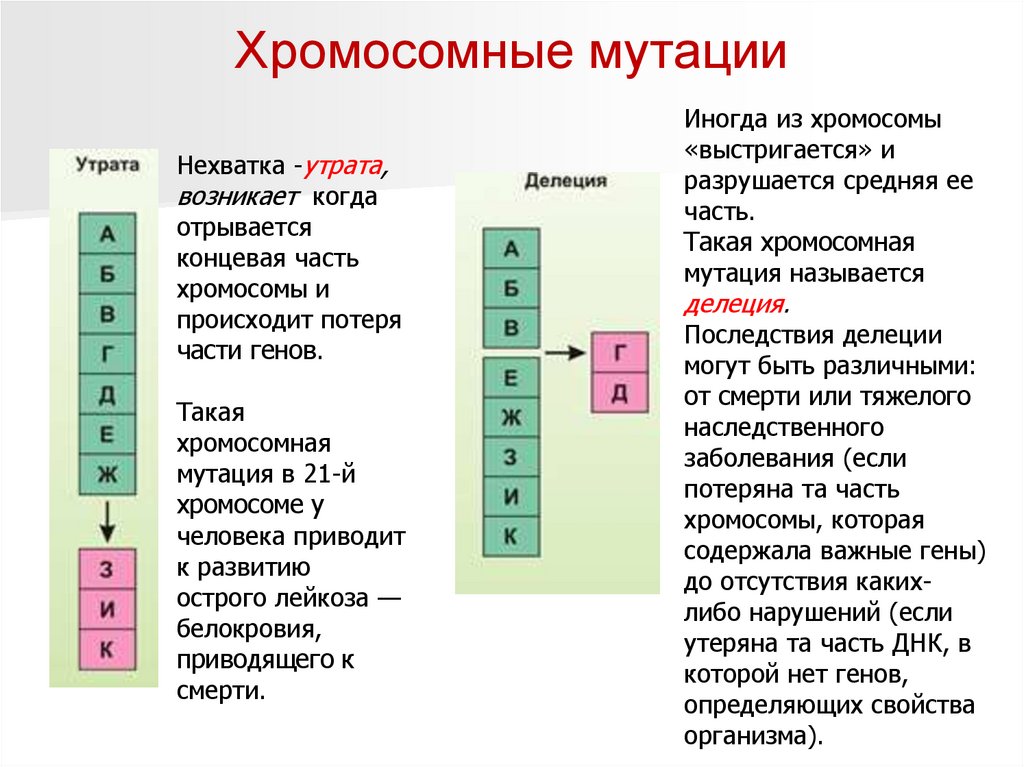
Хромосомные мутацииНехватка -утрата,
возникает когда
отрывается
концевая часть
хромосомы и
происходит потеря
части генов.
Такая
хромосомная
мутация в 21-й
хромосоме у
человека приводит
к развитию
острого лейкоза —
белокровия,
приводящего к
смерти.
Иногда из хромосомы
«выстригается» и
разрушается средняя ее
часть.
Такая хромосомная
мутация называется
делеция.
Последствия делеции
могут быть различными:
от смерти или тяжелого
наследственного
заболевания (если
потеряна та часть
хромосомы, которая
содержала важные гены)
до отсутствия какихлибо нарушений (если
утеряна та часть ДНК, в
которой нет генов,
определяющих свойства
организма).
32.
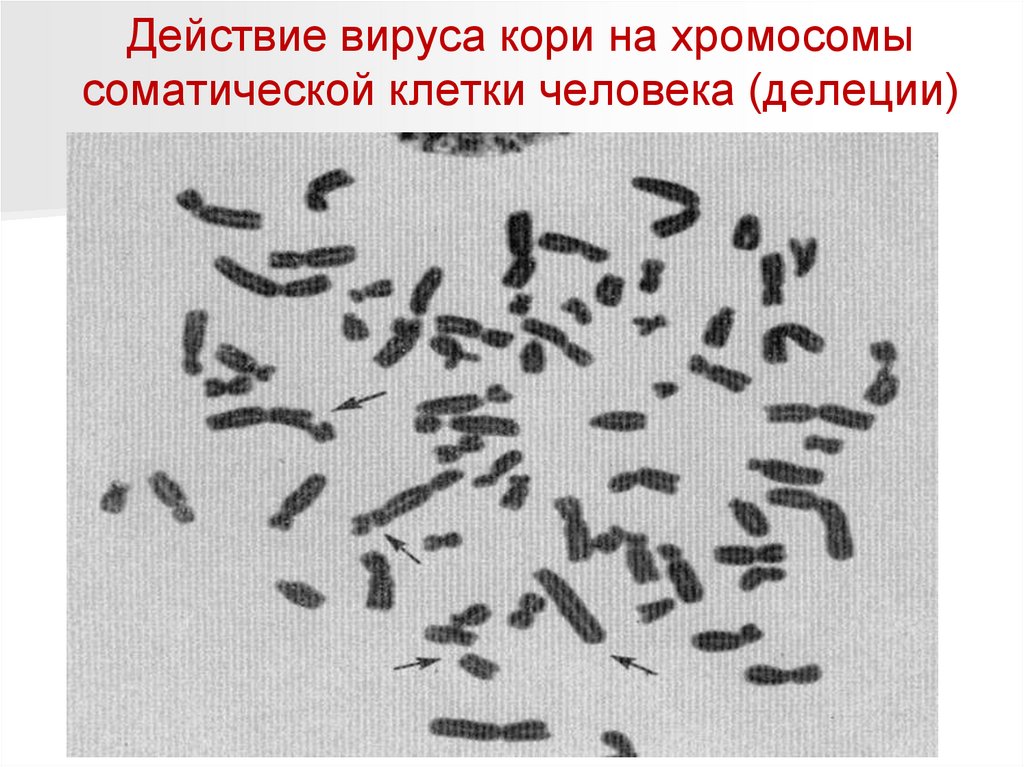
Действие вируса кори на хромосомысоматической клетки человека (делеции)
33.
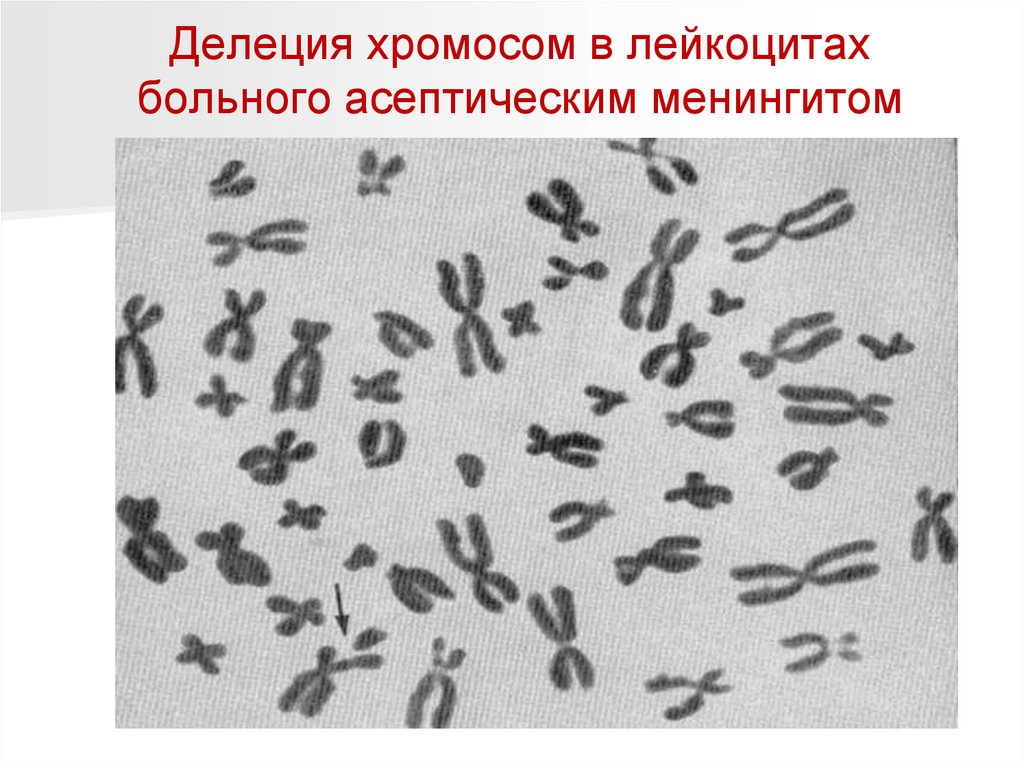
Делеция хромосом в лейкоцитахбольного асептическим менингитом
34.
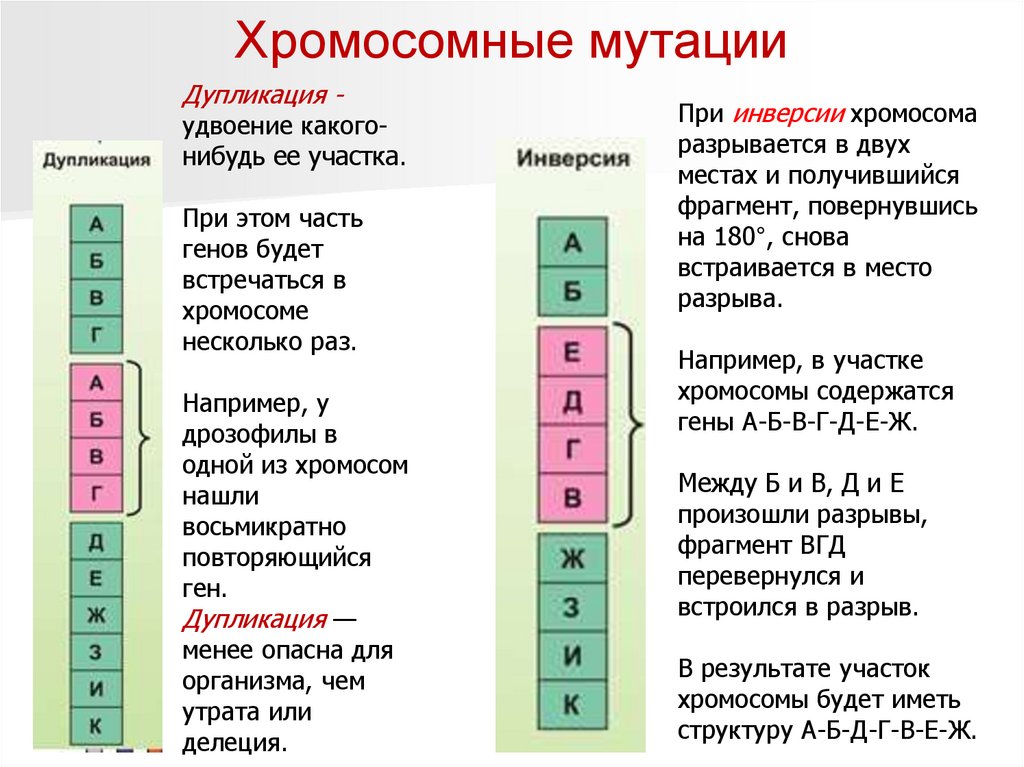
Хромосомные мутацииДупликация -
удвоение какогонибудь ее участка.
При этом часть
генов будет
встречаться в
хромосоме
несколько раз.
Например, у
дрозофилы в
одной из хромосом
нашли
восьмикратно
повторяющийся
ген.
Дупликация —
менее опасна для
организма, чем
утрата или
делеция.
При инверсии хромосома
разрывается в двух
местах и получившийся
фрагмент, повернувшись
на 180°, снова
встраивается в место
разрыва.
Например, в участке
хромосомы содержатся
гены А-Б-В-Г-Д-Е-Ж.
Между Б и В, Д и Е
произошли разрывы,
фрагмент ВГД
перевернулся и
встроился в разрыв.
В результате участок
хромосомы будет иметь
структуру А-Б-Д-Г-В-Е-Ж.
35.
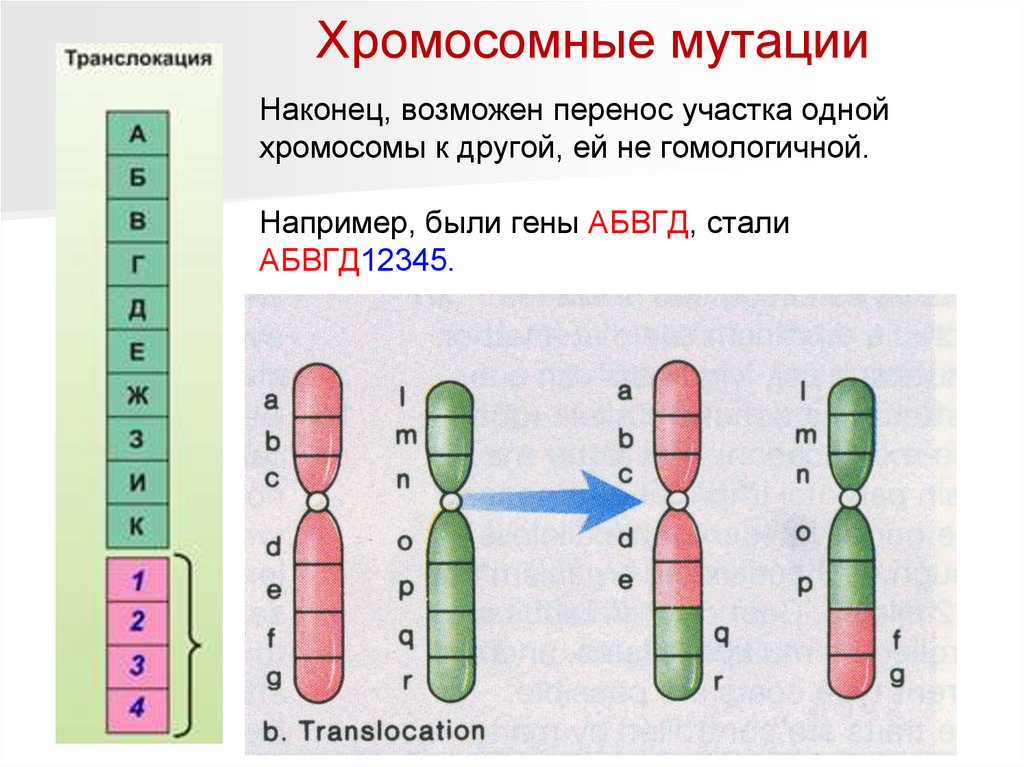
Хромосомные мутацииНаконец, возможен перенос участка одной
хромосомы к другой, ей не гомологичной.
Например, были гены АБВГД, стали
АБВГД12345.
36.
Робертсоновские транслокации- центрическое
соединения
между собой
длинных плеч
акроцентрических
хромосом.
Так возникает транслокационный синдром болезни
Дауна (транслокация лишней хромосомы 21 на одну из
хромосом группы D или G).
37.
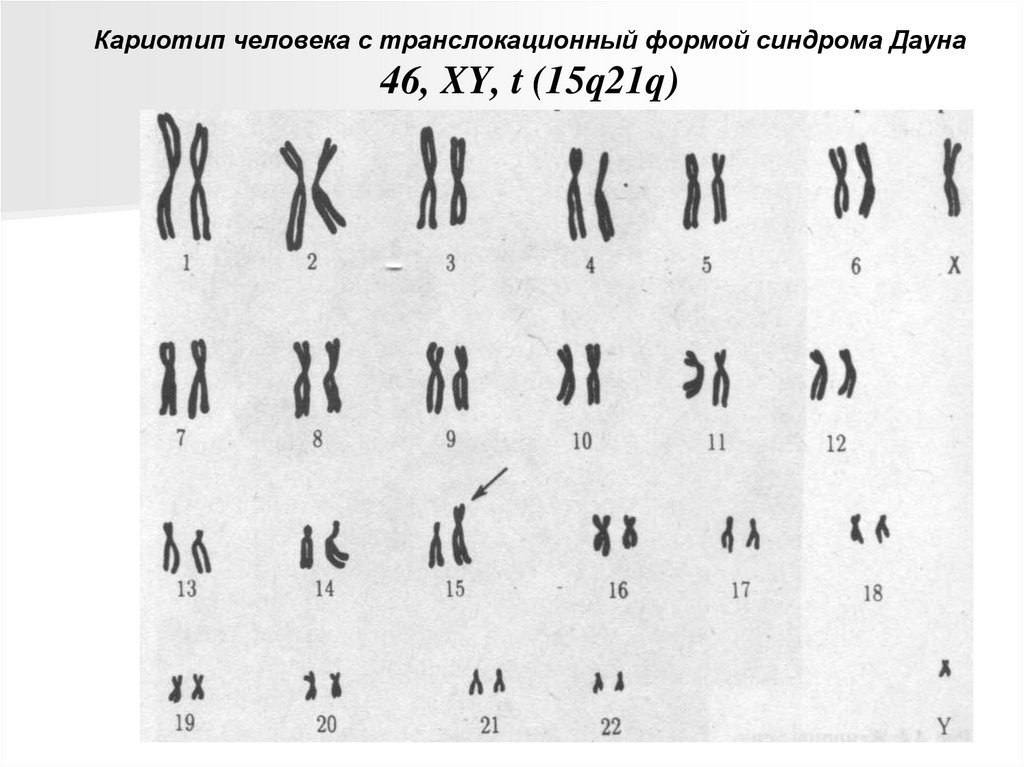
Кариотип человека с транслокационный формой синдрома Дауна46, XY, t (15q21q)
38.
Хромосомные мутации– В результате перестроек могут получаться
аномальные хромосомы.
– Любая получившаяся хромосома, которая не
обладает центромерой (ацентрическая) или
имеет две центромеры (дицентрическая), не
будет успешно разделяться при митозе и
будет в конечном итоге утрачиваться.
– Хромосомы с одной центромерой могут
стабильно распространяться в ряду
последовательных митозов, даже если они
структурно аномальны.
39.
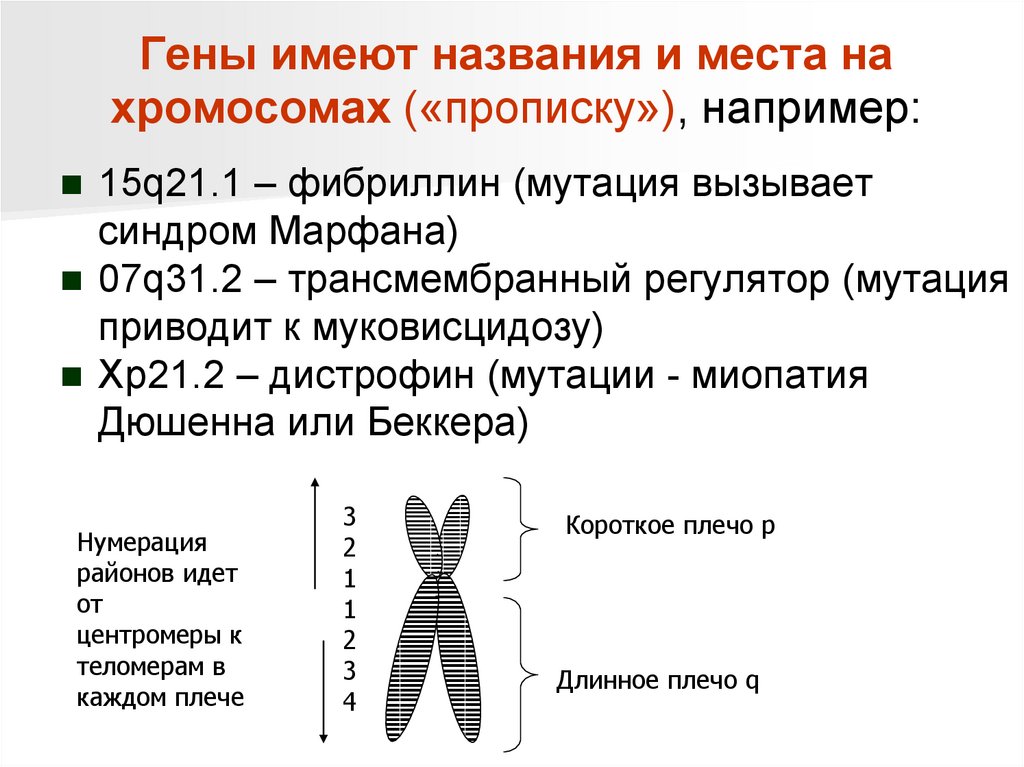
Гены имеют названия и места нахромосомах («прописку»), например:
15q21.1 – фибриллин (мутация вызывает
синдром Марфана)
07q31.2 – трансмембранный регулятор (мутация
приводит к муковисцидозу)
Xp21.2 – дистрофин (мутации - миопатия
Дюшенна или Беккера)
Нумерация
районов идет
от
центромеры к
теломерам в
каждом плече
3
2
1
1
2
3
4
Короткое плечо p
Длинное плечо q
40.
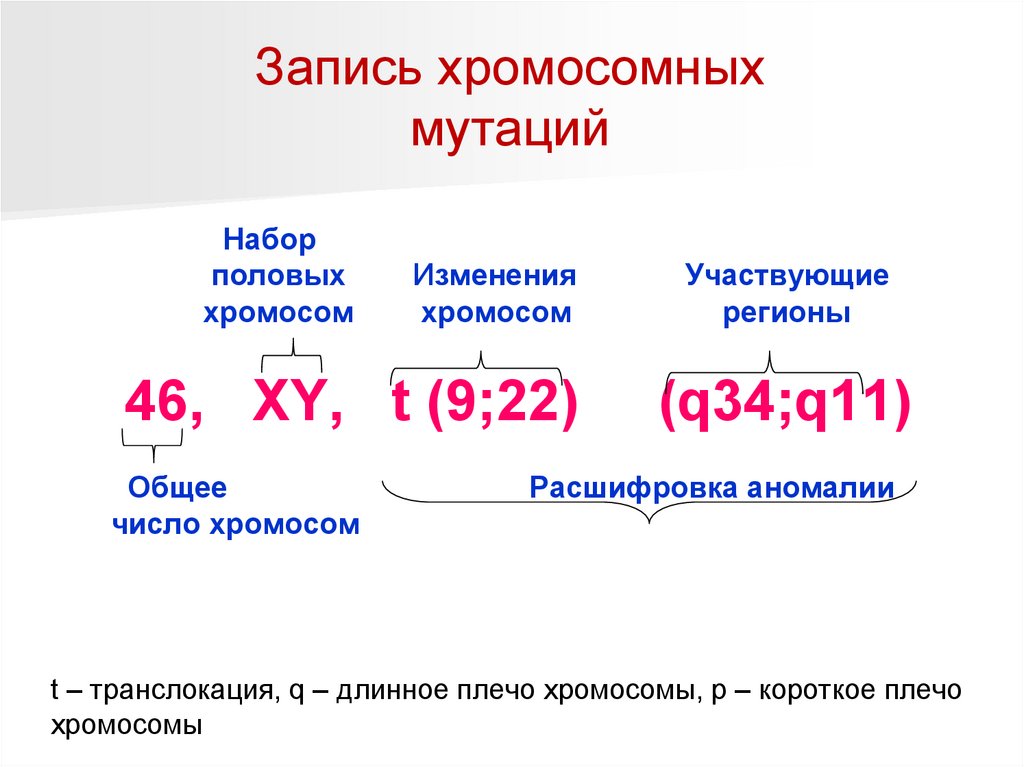
Запись хромосомныхмутаций
Набор
половых
хромосом
Изменения
хромосом
46, XY, t (9;22)
Общее
число хромосом
Участвующие
регионы
(q34;q11)
Расшифровка аномалии
t – транслокация, q – длинное плечо хромосомы, p – короткое плечо
хромосомы
41.
Генные или точковые мутации(трансгенации)
Генные, или точковые, мутации (трансгенации)
затрагивают структуру самого гена и являются
результатом изменения нуклеотидной
последовательности молекулы ДНК в определенном
участке хромосомы.
Изменение последовательности азотистых оснований в
данном гене воспроизводится при транскрипции в структуре
иРНК и приводит к изменению последовательности
аминокислот в полипептидной цепи.
Мутации изменяют участки ДНК разной длины. Наименьший
участок, изменение которого приводит к появлению мутации,
назван мутоном (может быть 1 нуклеотид)
42.
Типы генных мутацийСуществуют разные типы генных мутаций, связанных с
добавлением, выпадением или перестановкой нуклеотидов
в гене.
Это:
– Дупликации,
– Вставки лишней пары нуклеотидов,
– Делеции (выпадение пары нуклеотидов),
– Инверсии,
– Замены пар нуклеотидов.
Большая часть генных мутаций фенотипически не
проявляется (поскольку они рецессивны), однако известен
ряд случаев, когда изменение лишь одного основания в
определенном гене оказывает глубокое влияние на
фенотип.
43.
Замены нуклеотидовне обязательно ведут к
изменению смысла генетической
информации!
Миссенс
(missense)
изменяется
аминокислота в белке;
Сайлент
(silent) - аминокислота не
меняется;
Нонсенс (nonsense) - вместо кодона для
аминокислоты появляется стоп-кодон,
образуется осколочный белок.
44.
Генные мутацииМиссенс мутация. Пример – серповидно-клеточная анемия.
Замена пары нуклеотидов привела к
замене аминокислоты в белке, т.е.
изменилась первичная структура, что
повлекло изменение вторичной,
третичной и четвертичной и формы
эритроцитов.
Анемия вызывает физическую слабость,
нарушения деятельности сердца и почек и может
привести к ранней смерти людей, гомозиготных
по мутантному аллелю.
45.
Генные мутацииНонсенс мутация может возникнуть
как в
результате замены нуклеотида, так и при
сдвиге
рамки
считывания.
Пример:
появление
группы
крови
0.
У людей с данной группой крови в гене
произошло
выпадение
(делеция) одного
нуклеотида – в результате возник стоп-кодон.
Синтезируется короткий и неактивный белокфермент.
46.
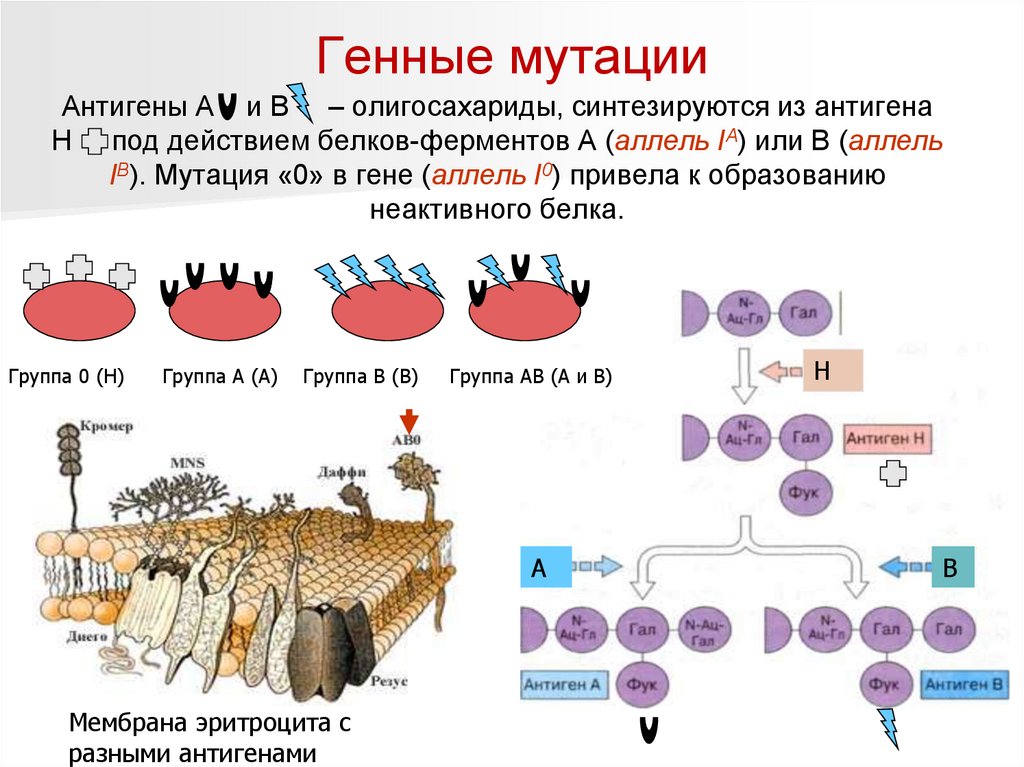
Генные мутацииАнтигены А и В – олигосахариды, синтезируются из антигена
Н под действием белков-ферментов А (аллель IA) или В (аллель
IВ). Мутация «0» в гене (аллель I0) привела к образованию
неактивного белка.
Группа 0 (Н)
Группа А (А)
Группа В (В)
Группа АВ (А и В)
А
Мембрана эритроцита с
разными антигенами
Н
В
47.
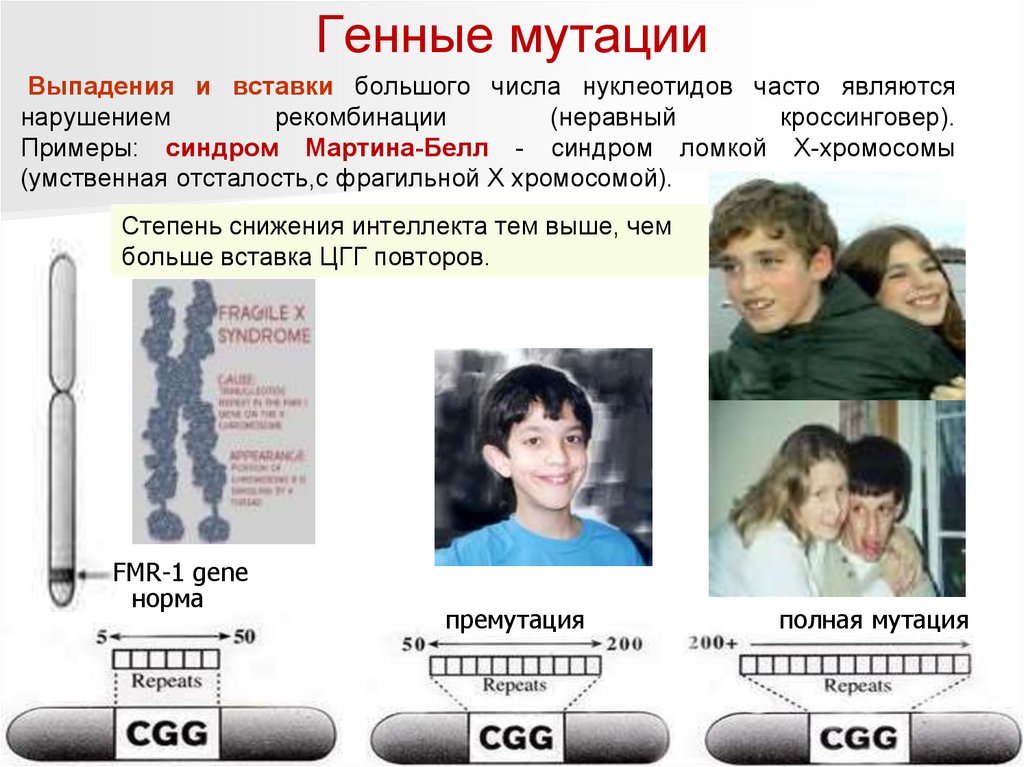
Генные мутацииВыпадения и вставки большого числа нуклеотидов часто являются
нарушением
рекомбинации
(неравный
кроссинговер).
Примеры: синдром Мартина-Белл - синдром ломкой Х-хромосомы
(умственная отсталость,с фрагильной Х хромосомой).
Степень снижения интеллекта тем выше, чем
больше вставка ЦГГ повторов.
FMR-1 gene
норма
премутация
полная мутация
48.
Генные мутации5. По локализации в гене:
Если мутация происходит:
в кодирующей части – синтез белка может измениться
качественно;
в регуляторной части – например, в промоторе – синтез
белка может измениться количественно
в интронах - произойдет нейтральная (сайлент) мутация
- ничего не будет.
6. По локализации в клетке:
Ядерные
Цитоплазматические
(немногочисленные, но тяжелые
митохондриальные болезни)
Митохондрии имеют
свою кольцевую ДНК
49.
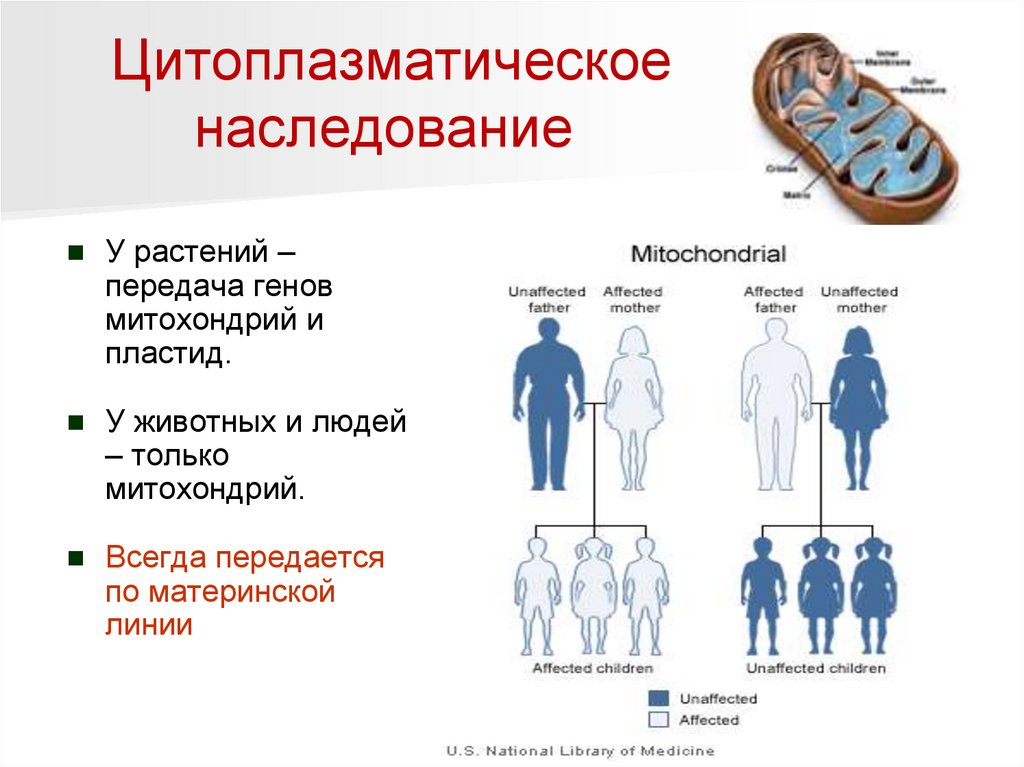
Цитоплазматическоенаследование
У растений –
передача генов
митохондрий и
пластид.
У животных и людей
– только
митохондрий.
Всегда передается
по материнской
линии
50.
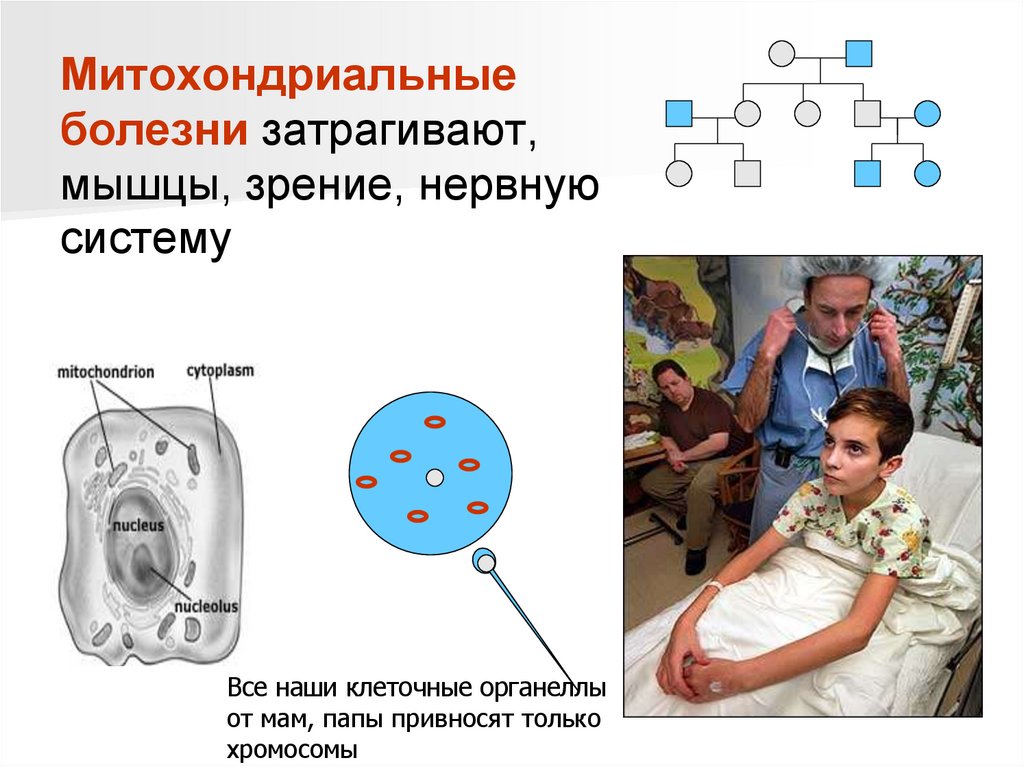
Митохондриальныеболезни затрагивают,
мышцы, зрение, нервную
систему
Все наши клеточные органеллы
от мам, папы привносят только
хромосомы
51.
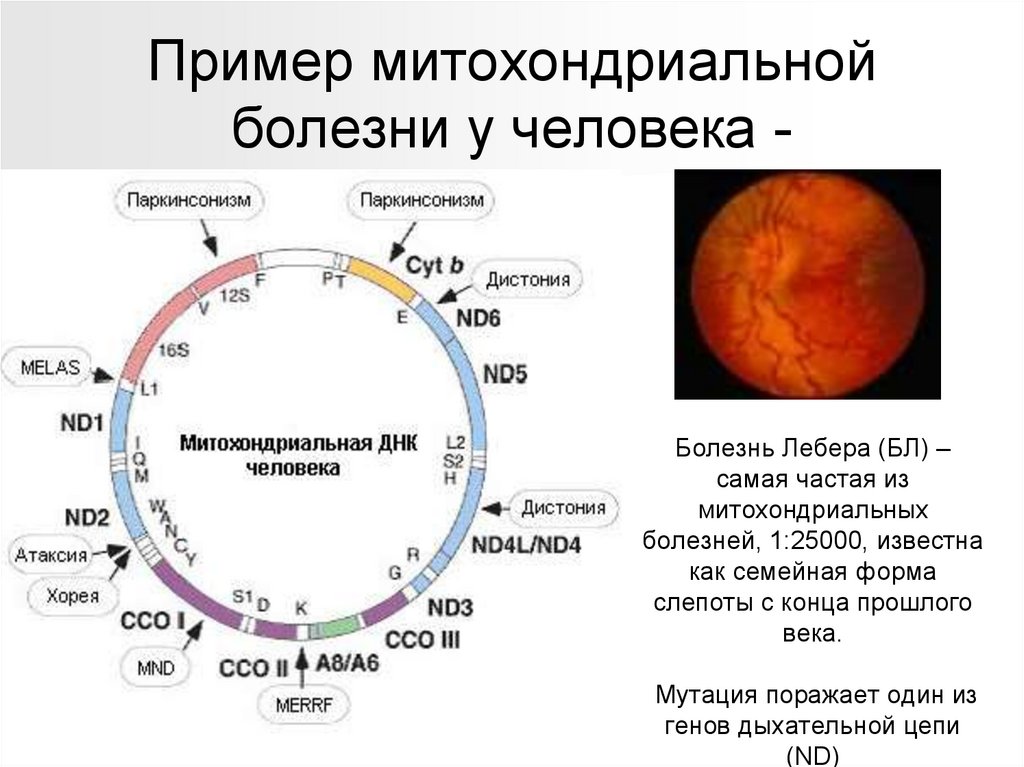
Пример митохондриальнойболезни у человека -
Болезнь Лебера (БЛ) –
самая частая из
митохондриальных
болезней, 1:25000, известна
как семейная форма
слепоты с конца прошлого
века.
Мутация поражает один из
генов дыхательной цепи
(ND)
52.
Но совсем без мутаций нельзя!вред для
особи
польза для
эволюции
53.
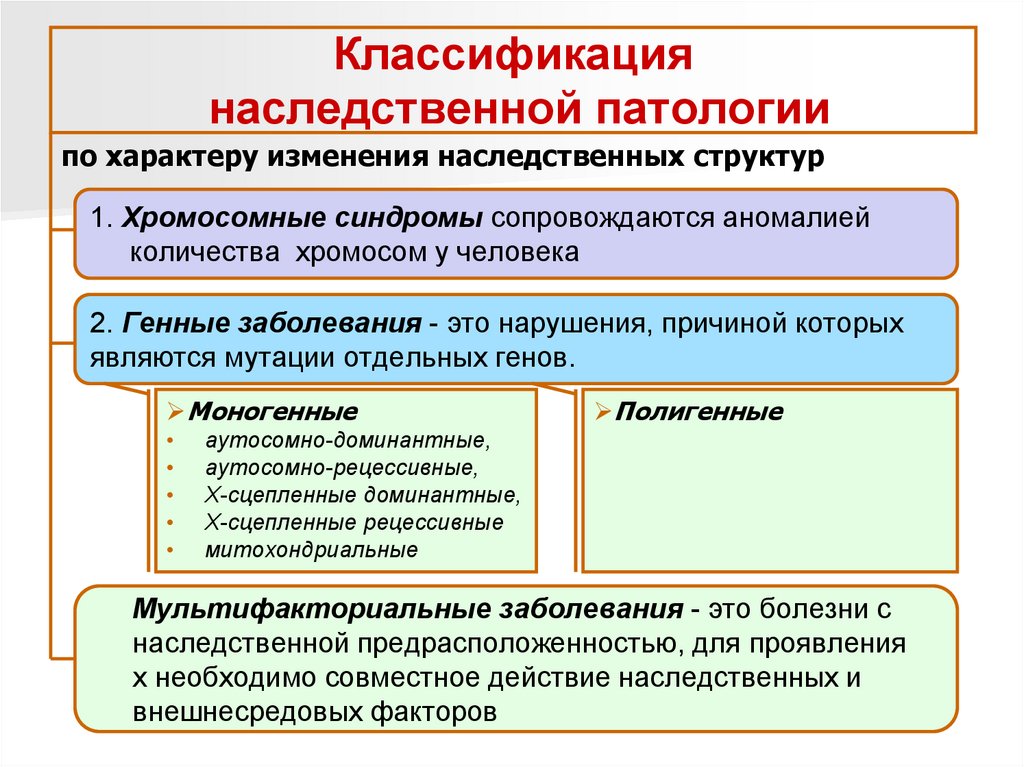
Классификациянаследственной патологии
по характеру изменения наследственных структур
1. Хромосомные синдромы сопровождаются аномалией
количества хромосом у человека
2. Генные заболевания - это нарушения, причиной которых
являются мутации отдельных генов.
Моногенные
Полигенные
аутосомно-доминантные,
аутосомно-рецессивные,
Х-сцепленные доминантные,
Х-сцепленные рецессивные
митохондриальные
Мультифакториальные заболевания - это болезни с
наследственной предрасположенностью, для проявления
х необходимо совместное действие наследственных и
внешнесредовых факторов
54.
Наследственные заболевания имеютобщие клинические особенности:
1.
2.
3.
4.
5.
6.
Повторные случаи аналогичной патологии у членов
одной семьи.
Врожденный характер заболевания. Большинство
пороков развития и хромосомных синдромов
обнаруживаются уже при рождении ребенка.
Хроническое, рецидивирующее течение
заболевания, приводящее к постепенному
ухудшению состояния больного.
Одновременное поражение у человека нескольких
органов или систем.
Наличие специфических симптомов.
Устойчивость к наиболее распространенным
методам терапии.
55.
Генные болезни - разнородная группанаследственных заболеваний человека, обусловленная
генными мутациями.
Общая частота генных болезней в популяциях людей –
2 - 4%.
В настоящее время описано более 5 тысяч таких
наследственных болезней.
Генные болезни делятся на две большие группы:
• болезни с выясненным первичным биохимическим
дефектом - наследственные болезни обмена веществ,
биосинтеза белка, ферментов.
болезни с невыясненным первичным биохимическим
дефектом.
56.
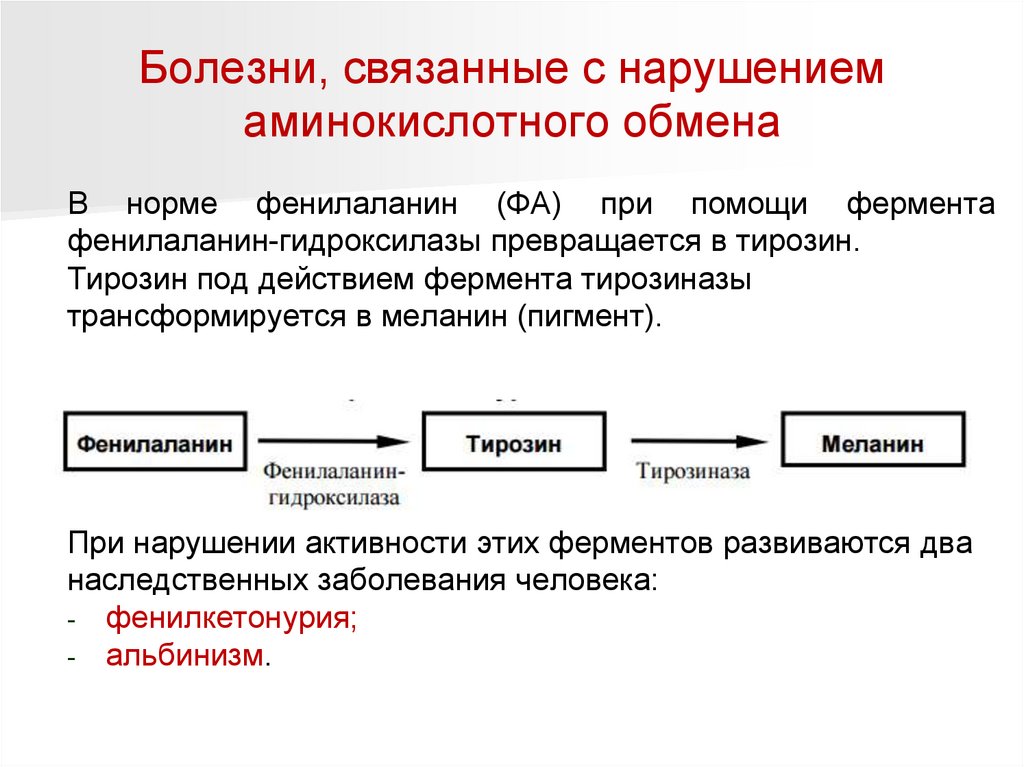
Болезни, связанные с нарушениемаминокислотного обмена
В норме фенилаланин (ФА) при помощи фермента
фенилаланин-гидроксилазы превращается в тирозин.
Тирозин под действием фермента тирозиназы
трансформируется в меланин (пигмент).
При нарушении активности этих ферментов развиваются два
наследственных заболевания человека:
- фенилкетонурия;
- альбинизм.
57.
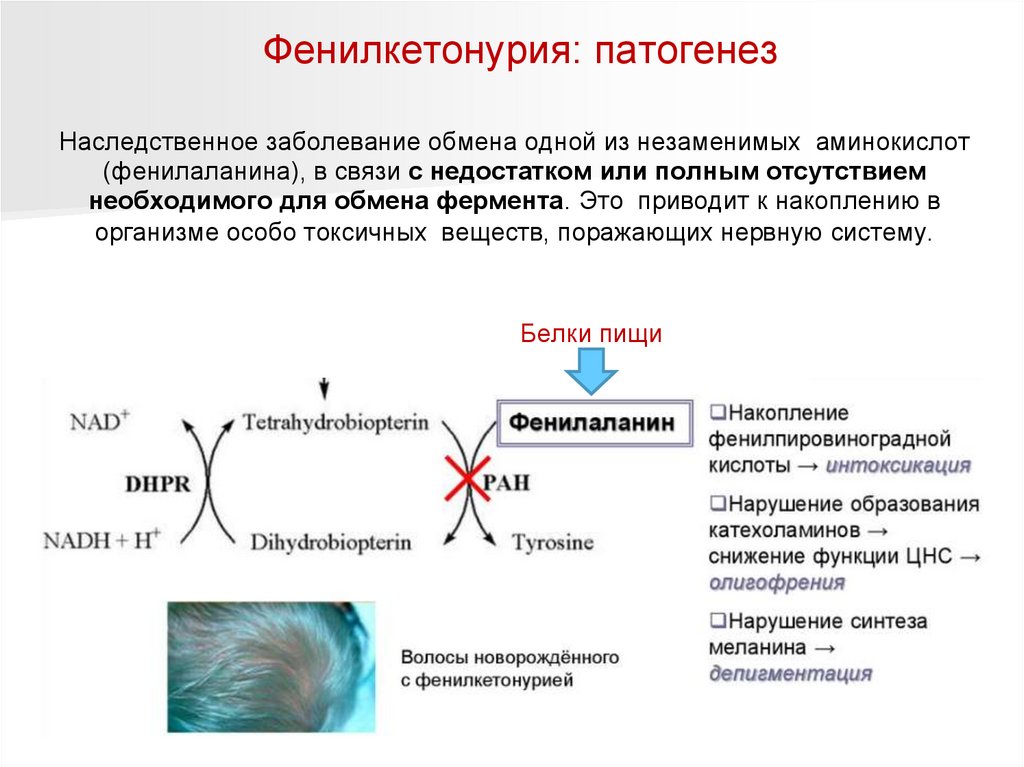
Фенилкетонурия (ФКУ, Болезнь Феллинга)Наследственное заболевание обмена одной из незаменимых аминокислот
(фенилаланина), в связи с недостатком или полным отсутствием
необходимого для обмена фермента. Это
приводит к накоплению в
организме особо токсичных веществ, поражающих нервную систему.
ФКУ наследуется по аутосомно-рецессивному типу, встречается с
частотой 1:10 000.
В России по данным неонатального скрининга частота фенилкетонурии
составляет 1:7000 и колеблется по регионам от 1:4735 в Курской области до
1:18000 в Республике Тыва.
В Санкт-Петербурге частота ФКУ 1:7600 новорожденных, в Москве 1:6772.
По половому признаку соотношение одинаковое, однако большинство
мальчиков с фенилкетонурией погибают еще на первом году жизни.
58.
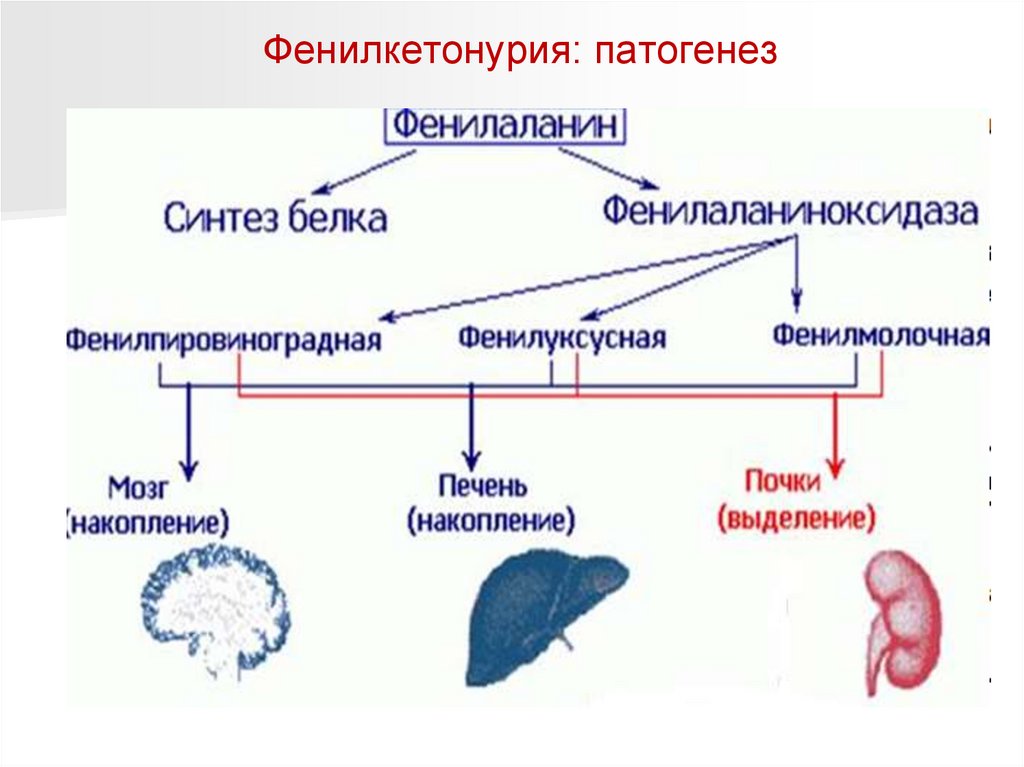
Фенилкетонурия: патогенезНаследственное заболевание обмена одной из незаменимых аминокислот
(фенилаланина), в связи с недостатком или полным отсутствием
необходимого для обмена фермента. Это приводит к накоплению в
организме особо токсичных веществ, поражающих нервную систему.
Белки пищи
59.
Фенилкетонурия: патогенез60.
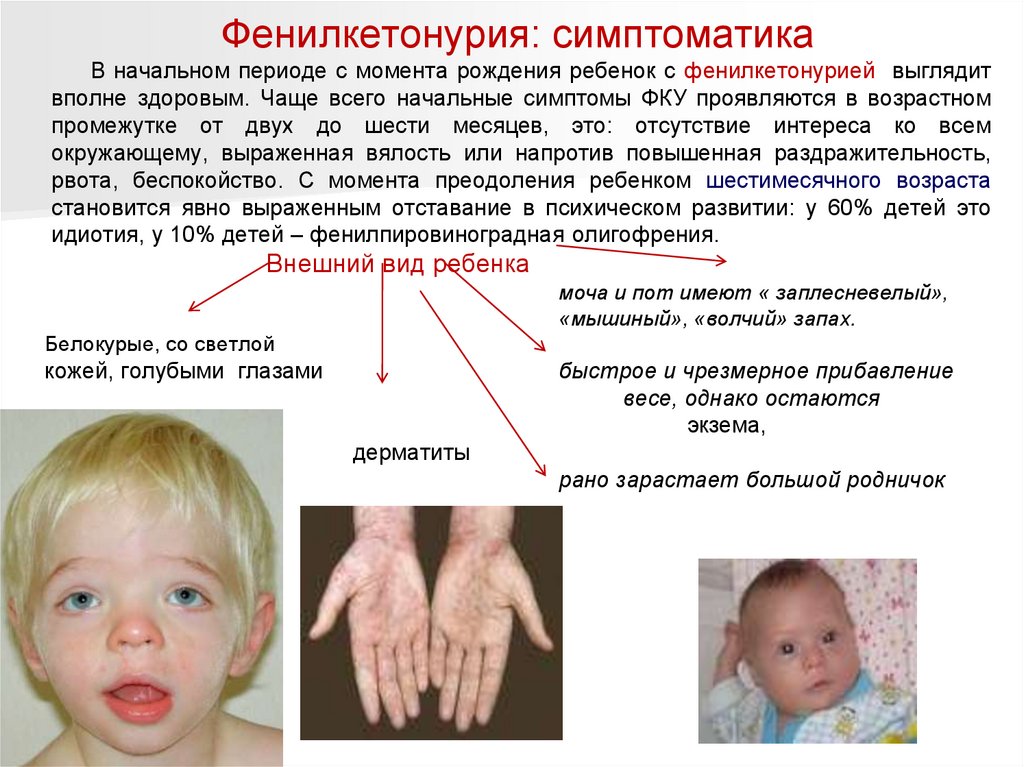
Фенилкетонурия: симптоматикаВ начальном периоде с момента рождения ребенок с фенилкетонурией выглядит
вполне здоровым. Чаще всего начальные симптомы ФКУ проявляются в возрастном
промежутке от двух до шести месяцев, это: отсутствие интереса ко всем
окружающему, выраженная вялость или напротив повышенная раздражительность,
рвота, беспокойство. С момента преодоления ребенком шестимесячного возраста
становится явно выраженным отставание в психическом развитии: у 60% детей это
идиотия, у 10% детей – фенилпировиноградная олигофрения.
Внешний вид ребенка
моча и пот имеют « заплесневелый»,
«мышиный», «волчий» запах.
Белокурые, со светлой
кожей, голубыми глазами
быстрое и чрезмерное прибавление
весе, однако остаются
экзема,
рыхлыми, вялыми.
дерматиты
рано зарастает большой родничок
61.
Фенилкетонурия: симптоматикаНаиболее характерные проявления фенилкетонурии:
• умственная отсталость ( 65% - глубокая, 31.8% - умеренная и 3.2% легкая);
• психические и неврологические расстройства, повышенная возбудимость
(в детском возрасте),
• Недоразвитие речи (ее или совсем нет, или есть
отдельные слова, которые больные не соотносят
с объектом), т.е. нарушено понимание речи
и звукопроизношение.
• специфическая поза и осанка при сидении
(«поза портного»), специфическая походка,
• стереотипные движения, необычное положение
конечностей, судороги, повышенные сухожильные рефлексы,
• дефектное формирование миелина,
• изменения кожных покровов, гипопигментация,
экзема, сухость; гипопигментированные волосы;
• микроцефалия,
• катаракта, светлые радужки, склеродермия,
• отчетливый «мышиный» запах тела,
• рвота в период новорожденности,
62.
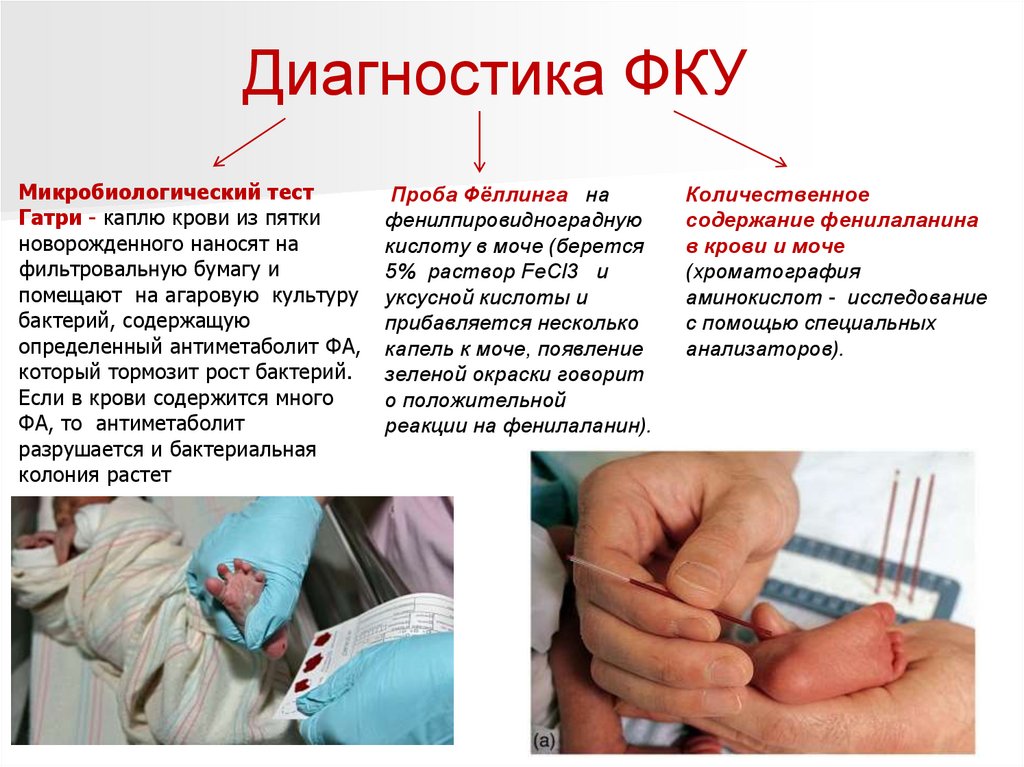
Диагностика ФКУМикробиологический тест
Гатри - каплю крови из пятки
новорожденного наносят на
фильтровальную бумагу и
помещают на агаровую культуру
бактерий, содержащую
определенный антиметаболит ФА,
который тормозит рост бактерий.
Если в крови содержится много
ФА, то антиметаболит
разрушается и бактериальная
колония растет
Проба Фёллинга на
фенилпировидноградную
кислоту в моче (берется
5% раствор FeCl3 и
уксусной кислоты и
прибавляется несколько
капель к моче, появление
зеленой окраски говорит
о положительной
реакции на фенилаланин).
Количественное
содержание фенилаланина
в крови и моче
(хроматография
аминокислот - исследование
с помощью специальных
анализаторов).
63.
Лечение больных ФКУСоблюдение диеты,
которой необходимо
придерживаться более
десяти лет после
постановки диагноза.
Нормы потребления ФА
зависят от возраста.
Ребенок до двух месяцев
может потреблять
не более 60 мг/кг массы ФА,
а дети старше шести лет –
не более 10-15 мг/кг.
Необходим ежедневный
подсчет уровня ФА,
получаемого с пищей,
учет белков, жиров, углеводов,
энергии, употребляемой
пациентом.
Введение прикорма у
детей с ФКУ начинают с
фруктовых и ягодных соков.
Лекарственное
лечение фенилкетонурии
предполагает прием
витаминов, препаратов
кальция, железа и фосфора.
Показан прием препаратов
для улучшения мозгового
кровообращения,
микроциркуляции и тканевого
обмена
занятия лечебной
физкультурой и массаж.
64.
АльбинизмНаследуется по аутосомно-рецессивному типу. Частота встречаемости 1: 5 000 – 1: 25 000.
У некоторых других народностей альбиносы встречаются чаще.
Так, при обследовании 14 292 негритянских детей в Нигерии среди них оказалось 5
альбиносов, что соответствует частоте около 1 на 3 000, а среди индейцев Панамы
(залив Сан-Блаз) частота составила 1 на 132.
При данной патологии нарушается активность тирозиназы.
Диагностика: клинический осмотр. Лечение не разработано
65.
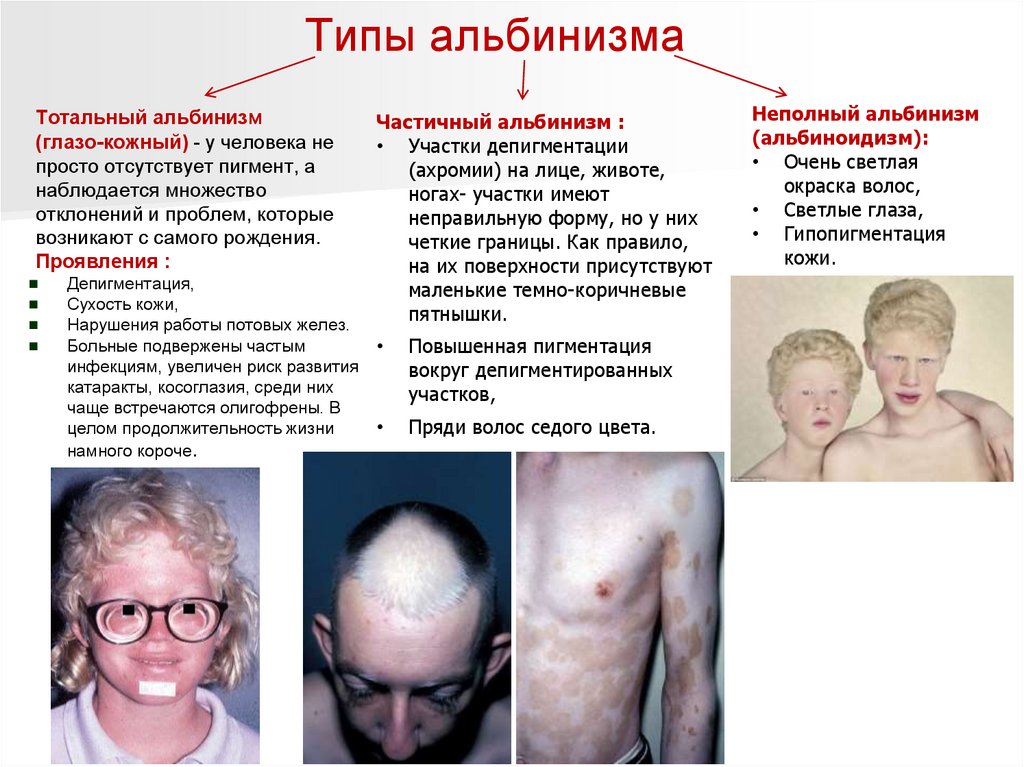
Типы альбинизмаТотальный альбинизм
(глазо-кожный) - у человека не
просто отсутствует пигмент, а
наблюдается множество
отклонений и проблем, которые
возникают с самого рождения.
Проявления :
Депигментация,
Сухость кожи,
Нарушения работы потовых желез.
Больные подвержены частым
инфекциям, увеличен риск развития
катаракты, косоглазия, среди них
чаще встречаются олигофрены. В
целом продолжительность жизни
намного короче.
Частичный альбинизм :
• Участки депигментации
(ахромии) на лице, животе,
ногах- участки имеют
неправильную форму, но у них
четкие границы. Как правило,
на их поверхности присутствуют
маленькие темно-коричневые
пятнышки.
Повышенная пигментация
вокруг депигментированных
участков,
Пряди волос седого цвета.
Неполный альбинизм
(альбиноидизм):
• Очень светлая
окраска волос,
• Светлые глаза,
• Гипопигментация
кожи.
66.
Болезни, связанные с нарушениемобмена углеводов.
• Углеводы входят в состав ряда биологически активных
веществ – гормонов, ферментов, мукополисахаридов,
выполняющих энергетическую и структурную функции.
• В результате нарушения углеводного обмена развивается
галактоземия и синдром Гурлера (мукополисахаридоз I типа)
67.
ГалактоземияДанная патология характеризуется нарушением естественной
способности организма расщеплять один из моносахаридов лактозы галактозу.
При данном дефекте не образуется один из важнейших ферментов,
влияющих на преобразование галактозы в глюкозу, следовательно, она
накапливается и оказывает токсическое действие на организм.
Отсутствие корректного и своевременного лечения может привести к
летальному исходу из-за церебральной или почечной недостаточности.
Вне зависимости от вида болезни она
передается по аутосомнорецессивному типу.
Частота встречаемости 1 : 100 000.
68.
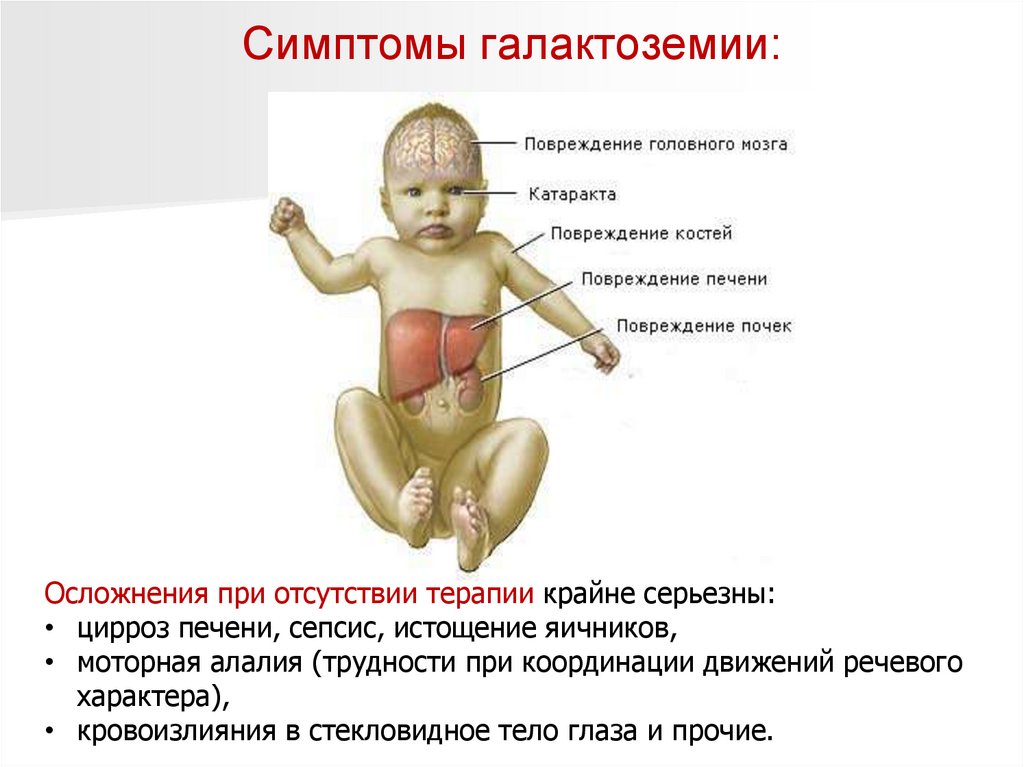
Симптомы галактоземии:Клинические симптомы галактоземии возникают у детей через
несколько дней после употребления молочной пищи.
Может появиться рвота и серьезное расстройство стула, после чего
наступает сильная интоксикация организма.
Для тяжелого течения характерны:
вялость, отказ от груди или бутылочки;
прогрессирующая гипотрофия (недостаточный прирост массы в
соответствии с возрастом и ростом малыша);
колики, обильное газообразование;
угасание характерных рефлексов;
прогрессирующая почечная недостаточность;
кровоизлияния на слизистых из-за нарушения нормального
свертывания крови;
развитие катаракты.
Развивается желтуха, увеличивается печень.
69.
Симптомы галактоземии:Осложнения при отсутствии терапии крайне серьезны:
• цирроз печени, сепсис, истощение яичников,
• моторная алалия (трудности при координации движений речевого
характера),
• кровоизлияния в стекловидное тело глаза и прочие.
70.
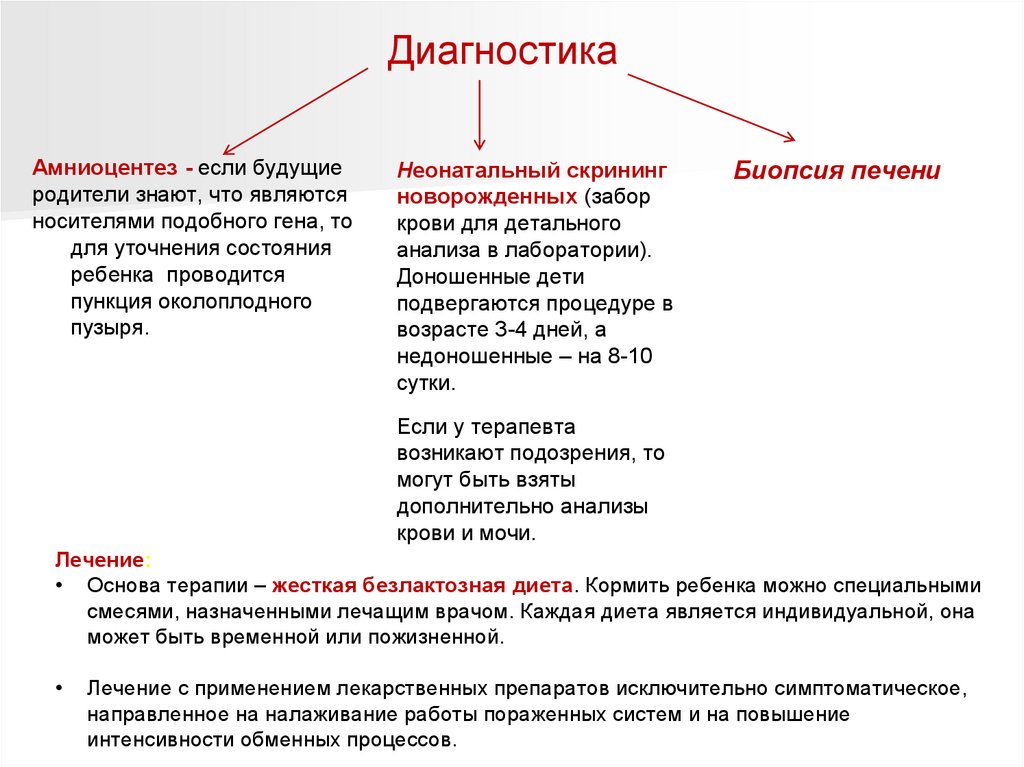
ДиагностикаАмниоцентез - если будущие
родители знают, что являются
носителями подобного гена, то
для уточнения состояния
ребенка проводится
пункция околоплодного
пузыря.
Неонатальный скрининг
новорожденных (забор
крови для детального
анализа в лаборатории).
Доношенные дети
подвергаются процедуре в
возрасте 3-4 дней, а
недоношенные – на 8-10
сутки.
Биопсия печени
Если у терапевта
возникают подозрения, то
могут быть взяты
дополнительно анализы
крови и мочи.
Лечение:
• Основа терапии – жесткая безлактозная диета. Кормить ребенка можно специальными
смесями, назначенными лечащим врачом. Каждая диета является индивидуальной, она
может быть временной или пожизненной.
Лечение с применением лекарственных препаратов исключительно симптоматическое,
направленное на налаживание работы пораженных систем и на повышение
интенсивности обменных процессов.
71.
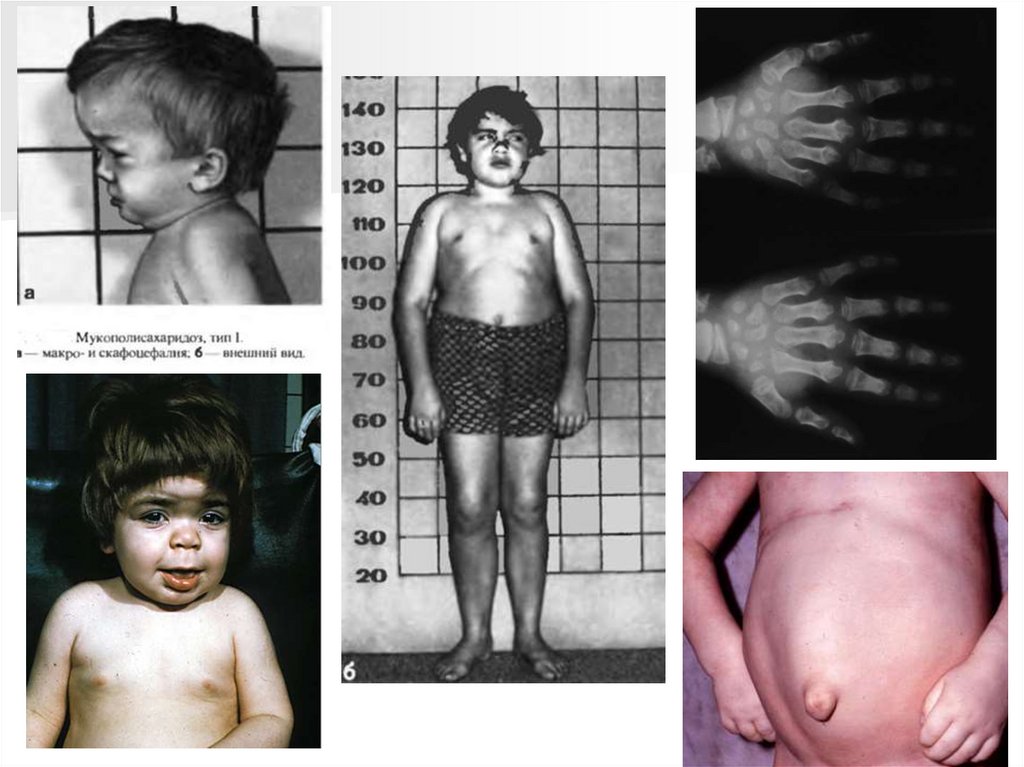
Мукополисахаридоз I типа (Синдром Гурлера)Наследственный дефект метаболизма гиалуроновой кислоты, связанный с
аутосомно-рецессивным типом наследования.
Частота встречаемости 1: 40 000.
Первые сведения о мукополисахаридозах появились в 1900 году. Еще
раньше это заболевание было известно под названием «гаргоилизм»,
поскольку внешне больные напоминали фигуры, украшавшие Собор
Парижской Богоматери.
Существует еще 15 типов
мукополисахиридозов.
72.
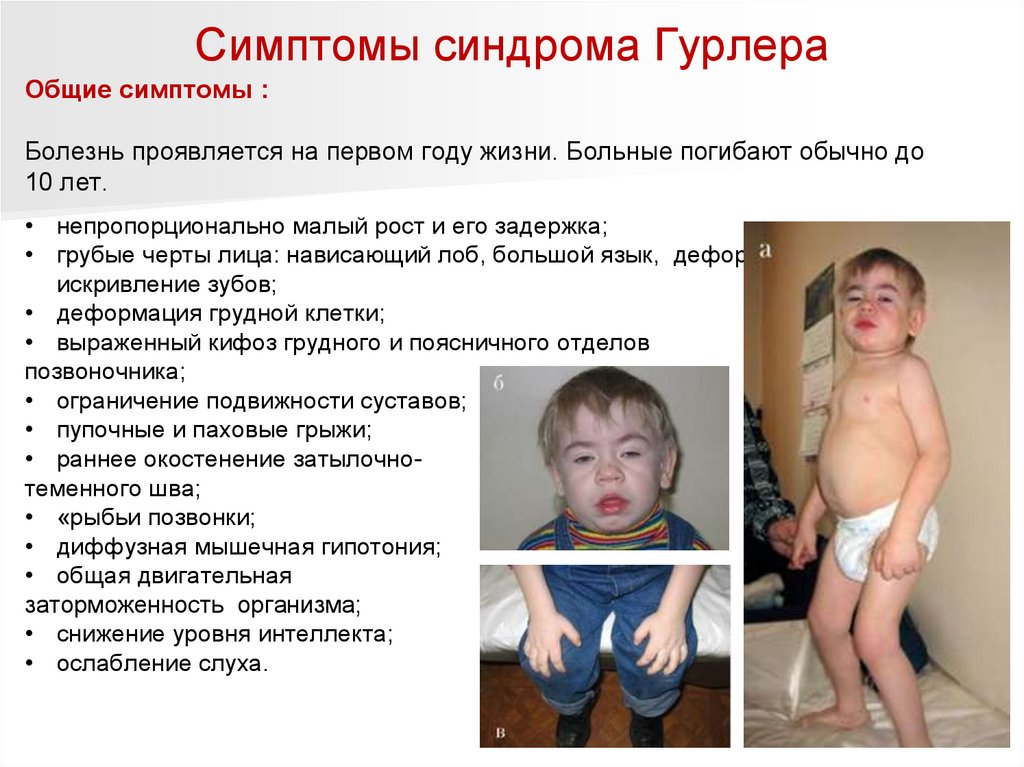
Симптомы синдрома ГурлераОбщие симптомы :
Болезнь проявляется на первом году жизни. Больные погибают обычно до
10 лет.
• непропорционально малый рост и его задержка;
• грубые черты лица: нависающий лоб, большой язык, деформация ушей и
искривление зубов;
• деформация грудной клетки;
• выраженный кифоз грудного и поясничного отделов
позвоночника;
• ограничение подвижности суставов;
• пупочные и паховые грыжи;
• раннее окостенение затылочнотеменного шва;
• «рыбьи позвонки;
• диффузная мышечная гипотония;
• общая двигательная
заторможенность организма;
• снижение уровня интеллекта;
• ослабление слуха.
73.
74.
Мукополисахаридоз 1 типа. ЛечениеПоскольку мукополисахаридоз 1 типа является генетическим
расстройством, то назначаемые терапии будут содержать как лечебные так
и паллиативные элементы.
- Фермент заместительная терапия лоранидазой может обеспечить
клинически важные преимущества, такие как будет происходить
улучшение легочной функции и сокращение избыточных углеводов,
хранящихся в органах.
- Хирургическая помощь: корректирующие операции могут быть
необходимы для пациентов с мукополисахаридозом 1 типа, которые имеют
контрактуры суставов, а также уродства рук и ног. Некоторым пациентам
может потребоваться пересадка роговицы, если проблемы со зрением
будут серьезными.
Мукополисахаридоз 1 типа. Прогноз
Течение заболевания злокачественное, приводит к инвалидизации больных
в течение нескольких лет.
75.
Болезни, связанные с нарушениями обменалипидов
Гликолипидозы – группа заболеваний, обусловленных нарушением распада
гликолипидов
(жироподобные
вещества,
содержащие
углеводы).
Гликолипидозы в основном наследуются по аутосомно-рецессивному типу
(болезнь
Гоше,
болезнь
Тея-Сакса
и
пр.).
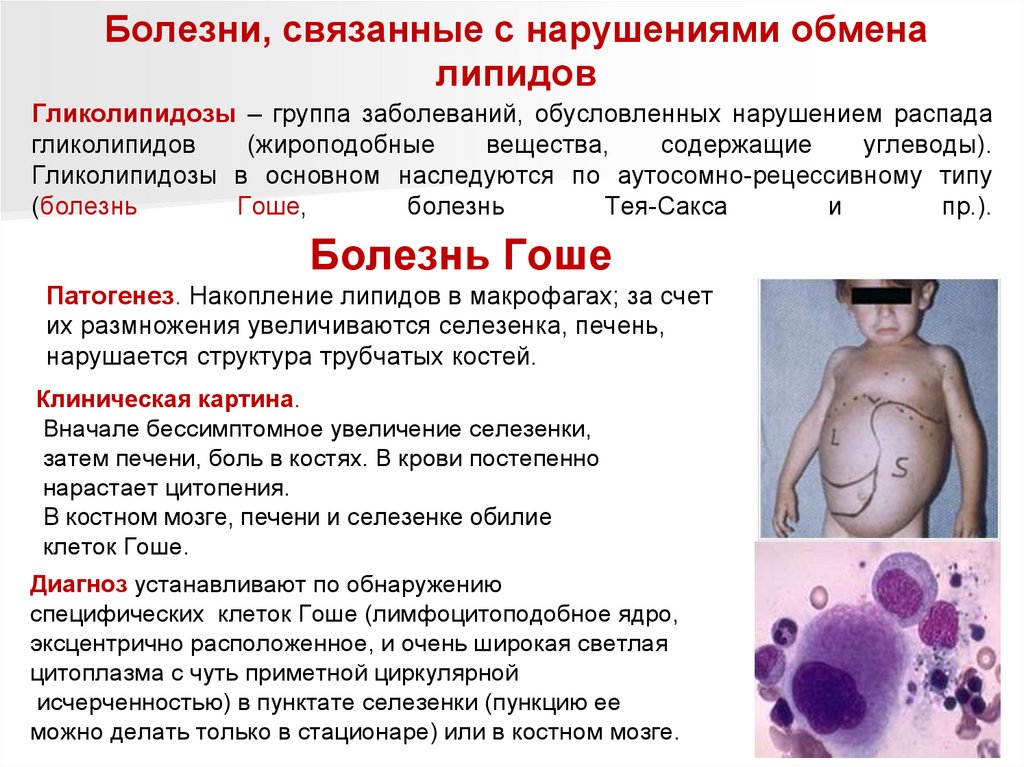
Болезнь Гоше
Патогенез. Накопление липидов в макрофагах; за счет
их размножения увеличиваются селезенка, печень,
нарушается структура трубчатых костей.
Клиническая картина.
Вначале бессимптомное увеличение селезенки,
затем печени, боль в костях. В крови постепенно
нарастает цитопения.
В костном мозге, печени и селезенке обилие
клеток Гоше.
Диагноз устанавливают по обнаружению
специфических клеток Гоше (лимфоцитоподобное ядро,
эксцентрично расположенное, и очень широкая светлая
цитоплазма с чуть приметной циркулярной
исчерченностью) в пунктате селезенки (пункцию ее
можно делать только в стационаре) или в костном мозге.
76.
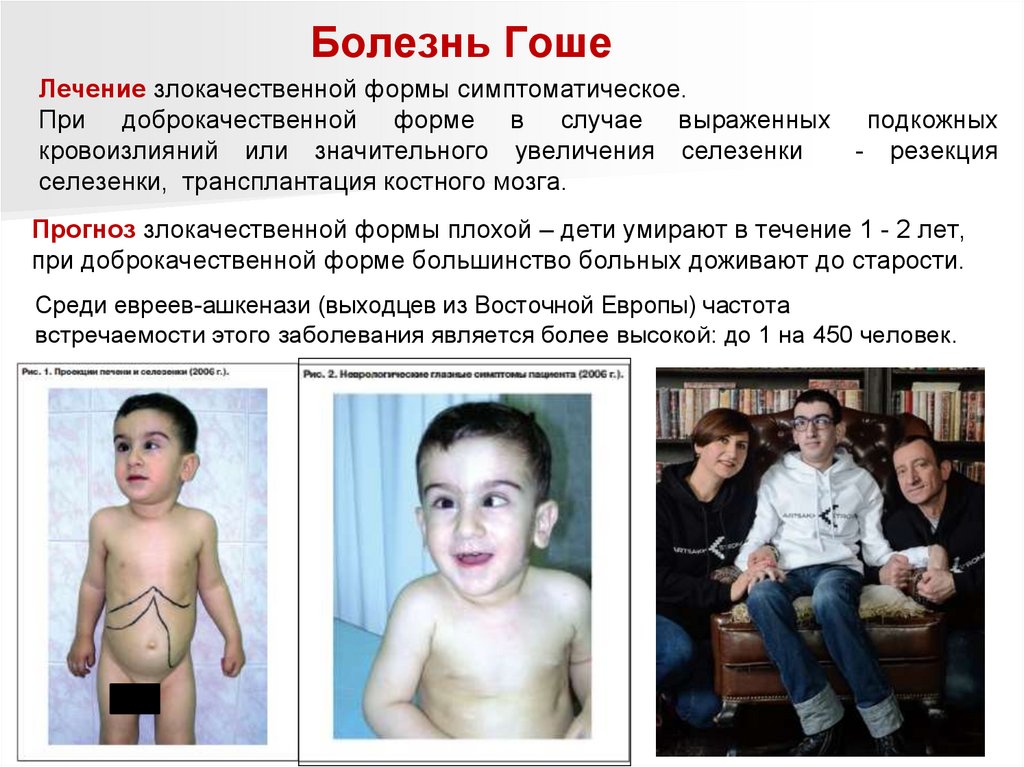
Болезнь ГошеЛечение злокачественной формы симптоматическое.
При доброкачественной форме в случае выраженных подкожных
кровоизлияний или значительного увеличения селезенки
- резекция
селезенки, трансплантация костного мозга.
Прогноз злокачественной формы плохой – дети умирают в течение 1 - 2 лет,
при доброкачественной форме большинство больных доживают до старости.
Среди евреев-ашкенази (выходцев из Восточной Европы) частота
встречаемости этого заболевания является более высокой: до 1 на 450 человек.
77.
Болезни, связанные с нарушением обмена пуринов ипиримидинов.
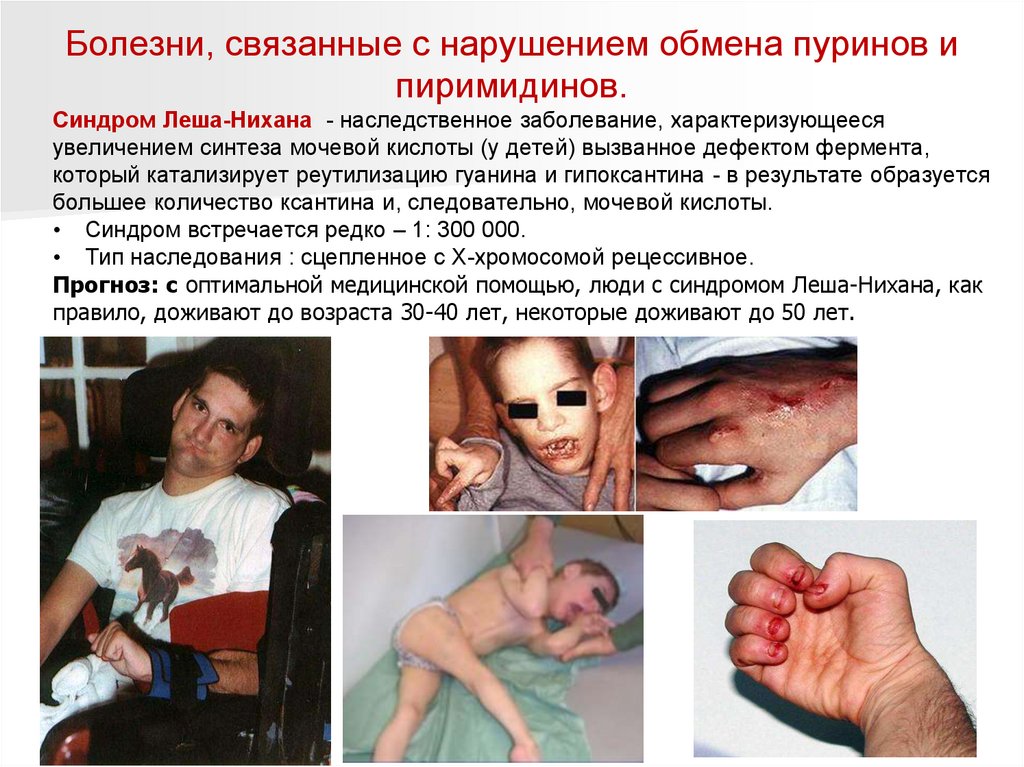
Синдром Леша-Нихана - наследственное заболевание, характеризующееся
увеличением синтеза мочевой кислоты (у детей) вызванное дефектом фермента,
который катализирует реутилизацию гуанина и гипоксантина - в результате образуется
большее количество ксантина и, следовательно, мочевой кислоты.
• Синдром встречается редко – 1: 300 000.
• Тип наследования : сцепленное с Х-хромосомой рецессивное.
Прогноз: с оптимальной медицинской помощью, люди с синдромом Леша-Нихана, как
правило, доживают до возраста 30-40 лет, некоторые доживают до 50 лет.
78.
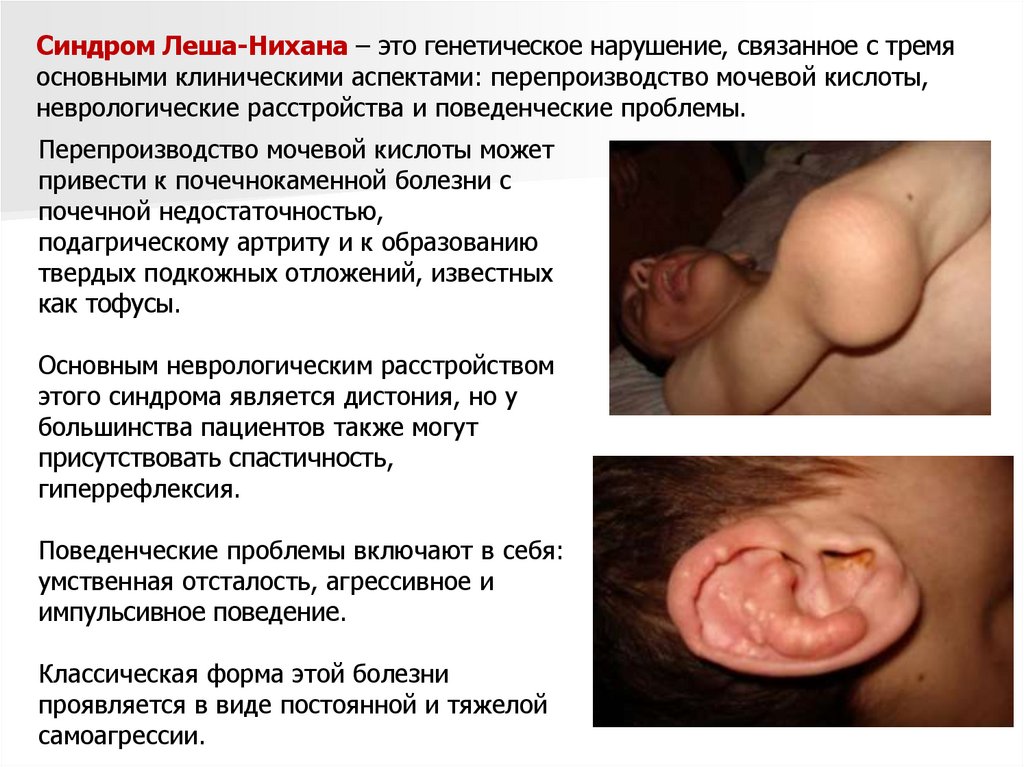
Синдром Леша-Нихана – это генетическое нарушение, связанное с тремяосновными клиническими аспектами: перепроизводство мочевой кислоты,
неврологические расстройства и поведенческие проблемы.
Перепроизводство мочевой кислоты может
привести к почечнокаменной болезни с
почечной недостаточностью,
подагрическому артриту и к образованию
твердых подкожных отложений, известных
как тофусы.
Основным неврологическим расстройством
этого синдрома является дистония, но у
большинства пациентов также могут
присутствовать спастичность,
гиперрефлексия.
Поведенческие проблемы включают в себя:
умственная отсталость, агрессивное и
импульсивное поведение.
Классическая форма этой болезни
проявляется в виде постоянной и тяжелой
самоагрессии.
79.
Нарушения минерального обменаБолезнь Вильсона-Коновалова - врожденное нарушение обмена меди в крови
человека. Аутосомно-рецессивный тип наследования. Частота встречаемости 1:35 000.
В области неврологии такое заболевание встречается крайне редко и страдают от него
чаще всего мужчины.
Начинает развиваться болезнь Вильсона-Коновалова в 4-х летнем возрасте. В этом
периоде симптомы проявляются в виде желтухи, увеличения размеров печени и
селезенки ребенка.
Неврологические изменения в организме начинают наблюдаться только спустя 10-20
лет.
Происходят следующие процессы:
• затрудняется выполнение тонких движений;
• проявляются дискоординаторные нарушения;
• меняется речь;
• развиваются психические нарушения.
Вскоре происходит повышение тонуса мышц лица,
туловища и всех конечностей, что в неврологии
становится причиной гипотомии, затруднения
глотания и измененной походки.
При развитии болезни Вильсона-Коновалова
полностью сохраняются сухожильные рефлексы
и не нарушаются функции тазовых органов.
Самым распространенным осложнением болезни Вильсона-Коновалова является
стремительное развитие слабоумия у больного. Кроме того, достаточно часто
происходят судороги, причиняющие боль и дискомфорт больному.
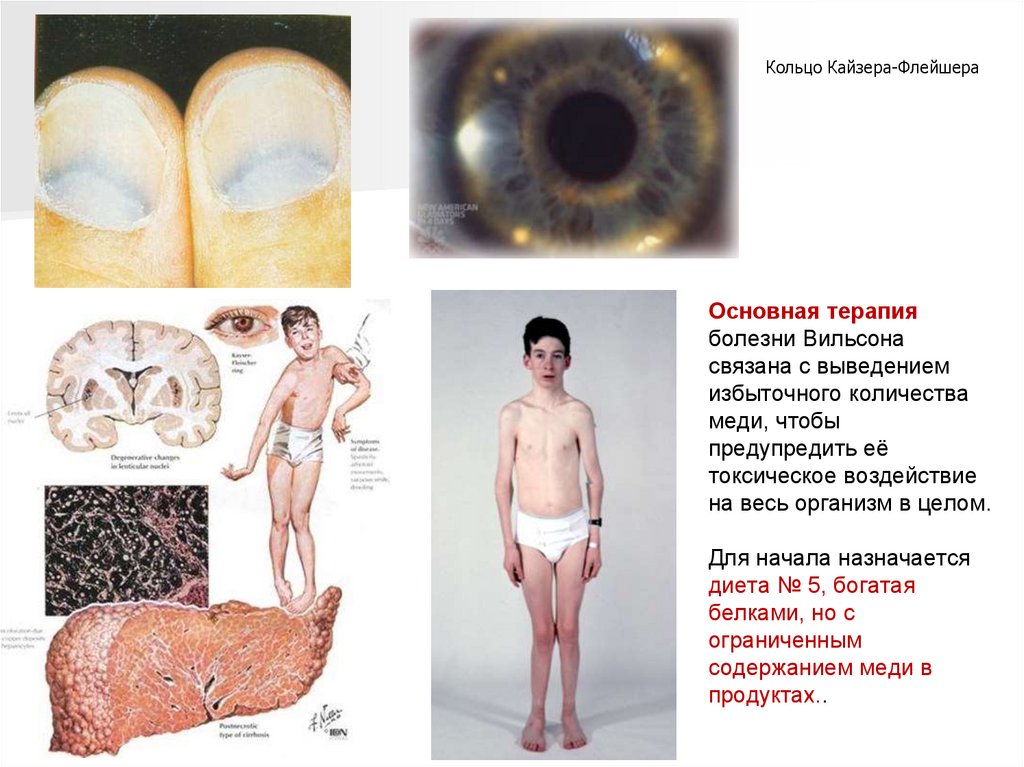
Характерным признаком является наличие голубой лунки ногтевого ложа.
80.
Кольцо Кайзера-ФлейшераОсновная терапия
болезни Вильсона
связана с выведением
избыточного количества
меди, чтобы
предупредить её
токсическое воздействие
на весь организм в целом.
Для начала назначается
диета № 5, богатая
белками, но с
ограниченным
содержанием меди в
продуктах..
81.
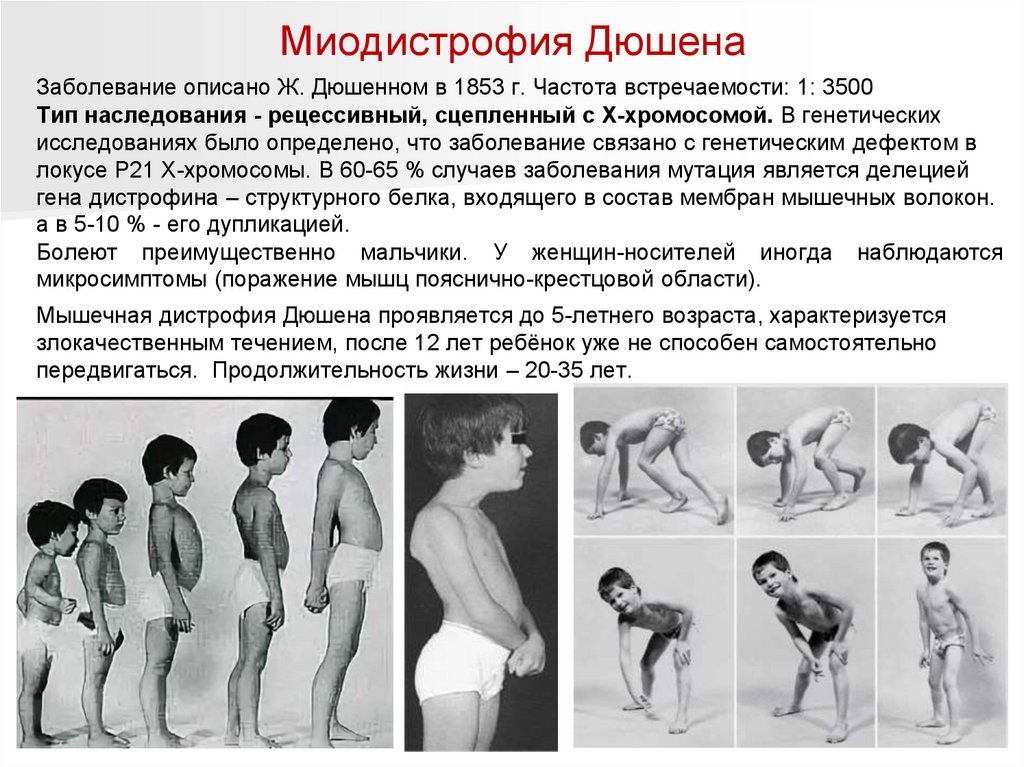
Миодистрофия ДюшенаЗаболевание описано Ж. Дюшенном в 1853 г. Частота встречаемости: 1: 3500
Тип наследования - рецессивный, сцепленный с Х-хромосомой. В генетических
исследованиях было определено, что заболевание связано с генетическим дефектом в
локусе Р21 Х-хромосомы. В 60-65 % случаев заболевания мутация является делецией
гена дистрофина – структурного белка, входящего в состав мембран мышечных волокон.
а в 5-10 % - его дупликацией.
Болеют преимущественно мальчики. У женщин-носителей иногда наблюдаются
микросимптомы (поражение мышц пояснично-крестцовой области).
Мышечная дистрофия Дюшена проявляется до 5-летнего возраста, характеризуется
злокачественным течением, после 12 лет ребёнок уже не способен самостоятельно
передвигаться. Продолжительность жизни – 20-35 лет.
82.
Миодистрофия ДюшенаОсновной симптом - мышечная слабость. С возрастом изменяется
последовательность поражения групп мышц больного: нарастающая слабость мышц
бедер и таза, позже верхнего плечевого пояса, спины и живота. Первым симптомом
мышечной дистрофии у детей является утолщение икроножных мышц. Мышцы при
пальпации плотные, болезненные.
Из горизонтального положения дети встают поэтапно с использованием
рук (взбирание лесенкой).
Утиная походка.
дыхательная недостаточность, кардиомиопатии,
уровень интеллекта понижен. Идет отставание в психомоторном развитии: такие
дети позже садятся, ползут, встают, идут.
Атрофии мышц приводят к развитию лордоза,
крыловидных лопаток
Деформация позвоночника
осиная
талия
Псевдогипертрофия икроножных
мышц.
Деформация стоп
83.
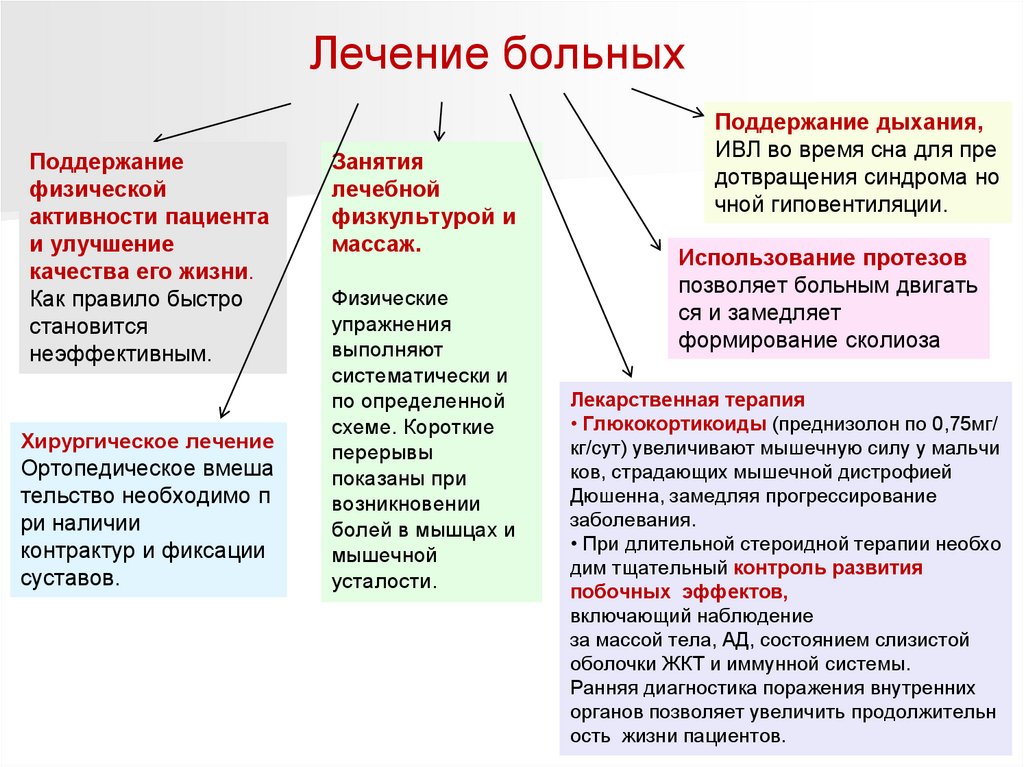
Лечение больныхПоддержание
физической
активности пациента
и улучшение
качества его жизни.
Как правило быстро
становится
неэффективным.
Хирургическое лечение
Ортопедическое вмеша
тельство необходимо п
ри наличии
контрактур и фиксации
суставов.
Занятия
лечебной
физкультурой и
массаж.
Физические
упражнения
выполняют
систематически и
по определенной
схеме. Короткие
перерывы
показаны при
возникновении
болей в мышцах и
мышечной
усталости.
Поддержание дыхания,
ИВЛ во время сна для пре
дотвращения синдрома но
чной гиповентиляции.
Использование протезов
позволяет больным двигать
ся и замедляет
формирование сколиоза
Лекарственная терапия
• Глюкокортикоиды (преднизолон по 0,75мг/
кг/сут) увеличивают мышечную силу у мальчи
ков, страдающих мышечной дистрофией
Дюшенна, замедляя прогрессирование
заболевания.
• При длительной стероидной терапии необхо
дим тщательный контроль развития
побочных эффектов,
включающий наблюдение
за массой тела, АД, состоянием слизистой
оболочки ЖКТ и иммунной системы.
Ранняя диагностика поражения внутренних
органов позволяет увеличить продолжительн
ость жизни пациентов.
84.
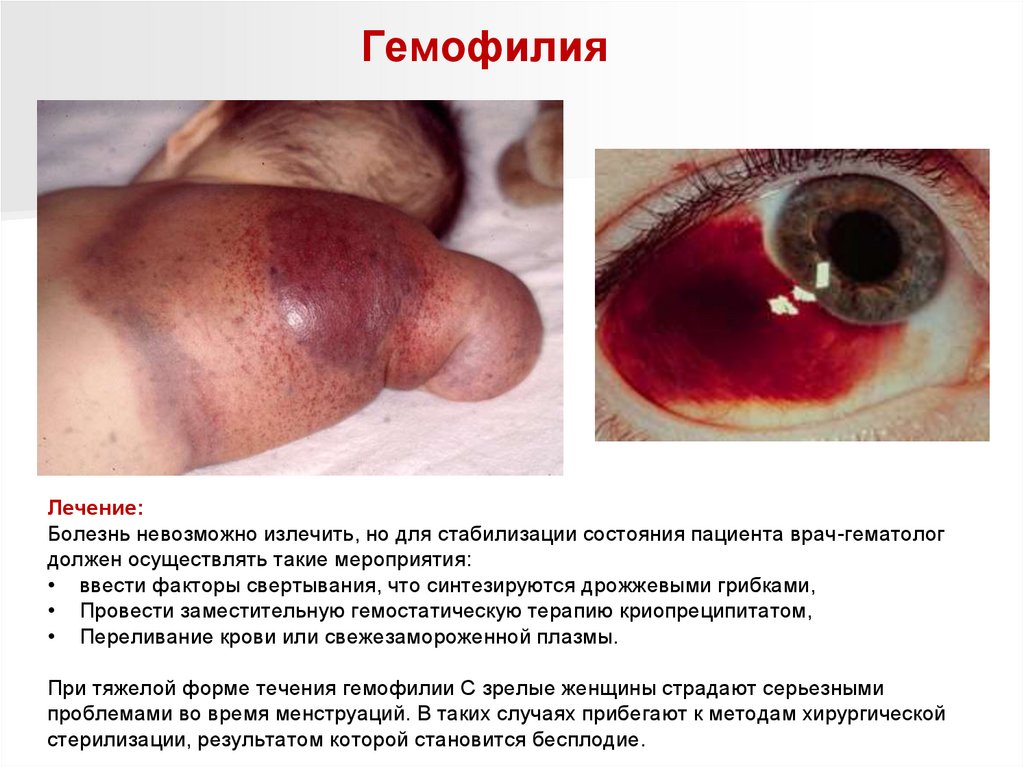
Нарушения свертывающей системы кровиГемофилии – наследственные заболевание, обусловленное мутацией генов,
отвечающих за формирование факторов свертывания крови в организме человека.
Гемофилия А и В - передаются сцепленно с Х-хромосомой, рецессивно.
Гемофилия С передается аутосомно-доминантно (страдают и женщины и мужчины).
Частота встречаемости гемофилии A составляет 1:10 000 мужчин, а гемофилии B —
1:25 000-1:55 000.
85.
Нарушения свертывающей системы кровиДиагностика базируется на :
- анализе семейного анамнеза (было ли наличие патологии близких или
дальних родственников),
- на жалобах (наличие длительных кровотечений, которые развиваются
после травмирования через несколько часов, частые кровоподтеки и
синяки на фоне ушибов незначительных),
- на общем осмотре пациента,
- лабораторной диагностике на свертываемые параметры.
Могут потребоваться консультация хирурга,
терапевта, генетика, травматолога-ортопеда.
Клинические проявления:
проявления кровоточивости в виде
кровоизлияний в суставы конечностей,
подкожные, внутримышечные и
межмышечные гематомы, обильные
кровотечения длительного характера при
травмах, после манипуляций.
86.
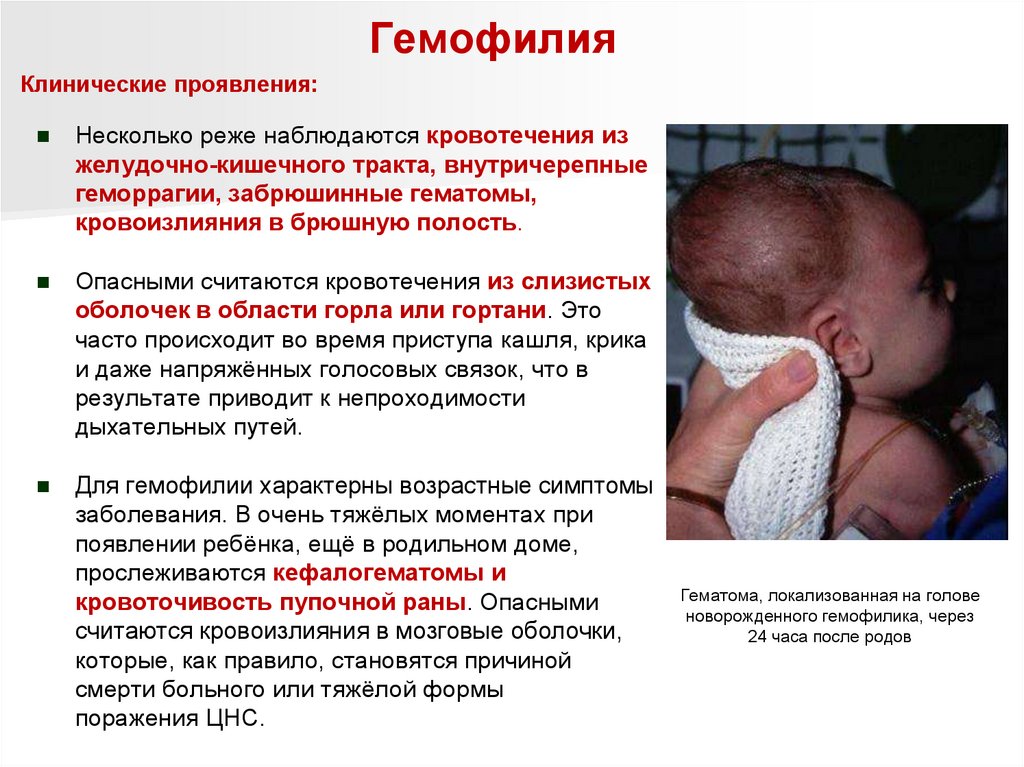
ГемофилияКлинические проявления:
Несколько реже наблюдаются кровотечения из
желудочно-кишечного тракта, внутричерепные
геморрагии, забрюшинные гематомы,
кровоизлияния в брюшную полость.
Опасными считаются кровотечения из слизистых
оболочек в области горла или гортани. Это
часто происходит во время приступа кашля, крика
и даже напряжённых голосовых связок, что в
результате приводит к непроходимости
дыхательных путей.
Для гемофилии характерны возрастные симптомы
заболевания. В очень тяжёлых моментах при
появлении ребёнка, ещё в родильном доме,
прослеживаются кефалогематомы и
кровоточивость пупочной раны. Опасными
считаются кровоизлияния в мозговые оболочки,
которые, как правило, становятся причиной
смерти больного или тяжёлой формы
поражения ЦНС.
Гематома, локализованная на голове
новорожденного гемофилика, через
24 часа после родов
87.
ГемофилияОчень часто первые симптомы гемофилии
обнаруживаются в связи с кровотечениями, которые
появляются после манипуляций в виде инъекций,
пункций, хирургических операций.
А вот гемартрозы возникают, как правило, без
видимых травм, но суставы становятся болезненными,
горячими, ригидными и согнутыми. Боль препятствует
движению.
При рентгенологическом исследовании изменения не
наблюдаются. Но после частых таких кровоизлияний
капсула сустава утолщается и изменяет свой цвет.
В дальнейшем в ней воспалительный процесс
увеличивается.
Обильные почечные кровотечения создают
серьёзные терапевтические проблемы, которые
диагностируют у больных гемофилией. Они
встречаются практически у 20% больных.
У больного проявляются приступы, связанные с
нарушением мочеиспускания и острыми болями
в поясничной области, в результате которой в
мочевыводящих путях образуются сгустки крови,
что может стать причиной гидронефроза.
гемартроз
88.
ГемофилияЛечение:
Болезнь невозможно излечить, но для стабилизации состояния пациента врач-гематолог
должен осуществлять такие мероприятия:
• ввести факторы свертывания, что синтезируются дрожжевыми грибками,
• Провести заместительную гемостатическую терапию криопреципитатом,
• Переливание крови или свежезамороженной плазмы.
При тяжелой форме течения гемофилии С зрелые женщины страдают серьезными
проблемами во время менструаций. В таких случаях прибегают к методам хирургической
стерилизации, результатом которой становится бесплодие.
89.
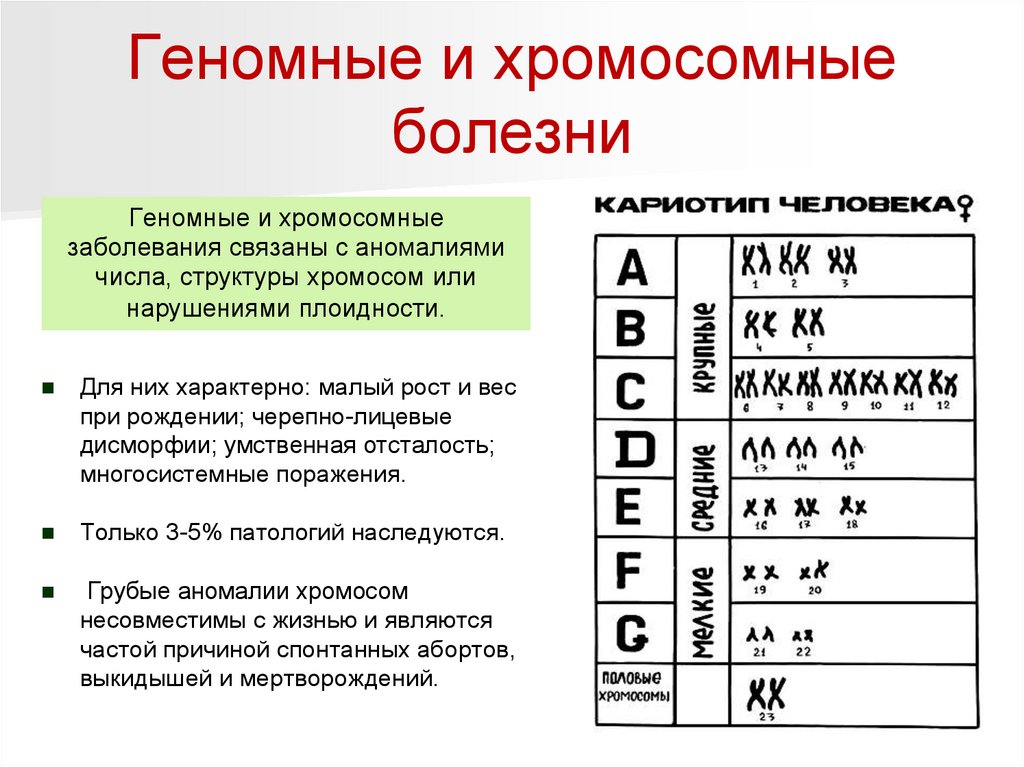
Геномные и хромосомныеболезни
Геномные и хромосомные
заболевания связаны с аномалиями
числа, структуры хромосом или
нарушениями плоидности.
Для них характерно: малый рост и вес
при рождении; черепно-лицевые
дисморфии; умственная отсталость;
многосистемные поражения.
Только 3-5% патологий наследуются.
Грубые аномалии хромосом
несовместимы с жизнью и являются
частой причиной спонтанных абортов,
выкидышей и мертворождений.
90.
91.
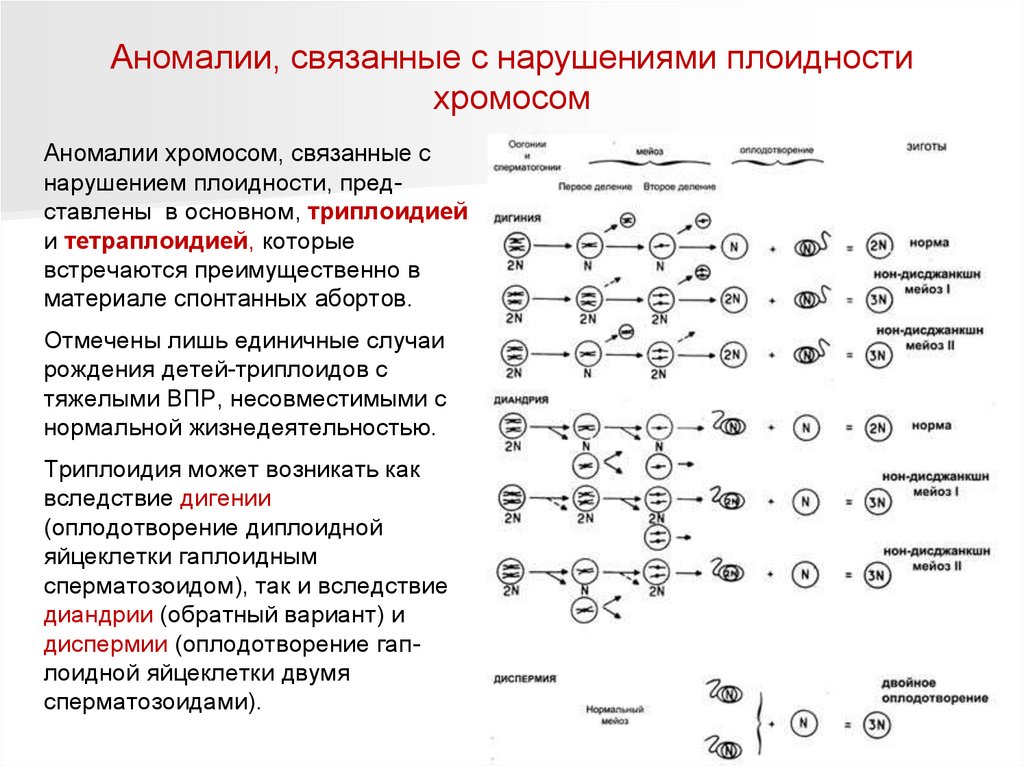
Аномалии, связанные с нарушениями плоидностихромосом
Аномалии хромосом, связанные с
нарушением плоидности, представлены в основном, триплоидией
и тетраплоидией, которые
встречаются преимущественно в
материале спонтанных абортов.
Отмечены лишь единичные случаи
рождения детей-триплоидов с
тяжелыми ВПР, несовместимыми с
нормальной жизнедеятельностью.
Триплоидия может возникать как
вследствие дигении
(оплодотворение диплоидной
яйцеклетки гаплоидным
сперматозоидом), так и вследствие
диандрии (обратный вариант) и
диспермии (оплодотворение гаплоидной яйцеклетки двумя
сперматозоидами).
92.
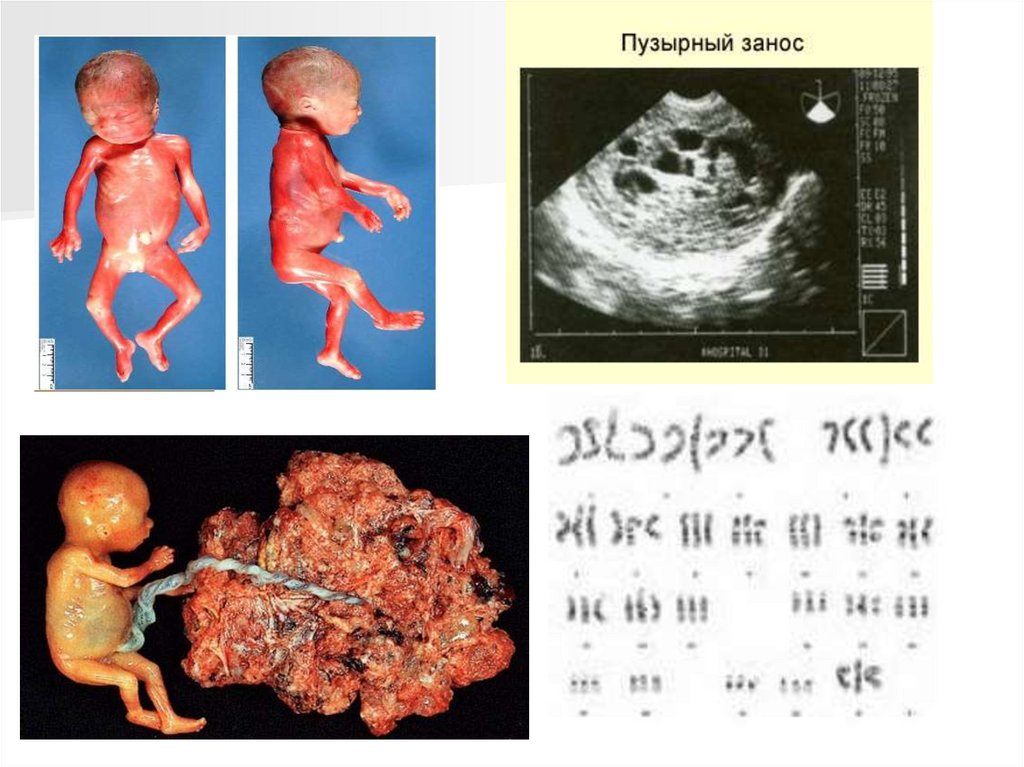
Три- и тетраплоидииНаличие трех наборов хромосом
приводит к локальному отечному
набуханию ворсин хориона с
гиперплазией трофобласта. Частота
встречаемости составляет 1% от
зачатий и 0,1-1 случай на 10 000
беременностей.
Триплоидия – одна из наиболее часто
встречающихся спонтанных аномалий
набора хромосом в эмбриогенезе
человека. Около 20% нарушений
хромосом у зародышей приходится
на триплоидию.
Синдром триплоидии (69, XXY) был
впервые обнаружен у человека в 60-х
годах. К настоящему времени
опубликовано около 60 случаев
триплоидии у детей, максимальная
продолжительность жизни которых
составляет 7 дней.
Тетраплоидия встречается крайне редко.
Из всех зародышей с хромосомными нарушениями обнаруживается лишь 5-6% случаев,
сопровождающихся серьезными пороками развития, такие зародыши редко вступают в
плодный период, погибая обычно в течение первых двух месяцев эмбриогенеза (описано 5
случаев рождения).
93.
94.
АнеуплоидияАнеуплоидии – изменение количества отдельных хромосом в кариотипе, не кратное
гаплоидному набору. В результате возникают особи с аномальным числом хромосом:
моносомики (2n-1) и полисомики (трисомики, тетрасомики и т.д.), когда одна из хромосом
может быть повторена трижды и более раз (2n+1,2…).
Чем меньше генов в хромосоме, тем вероятнее, что плод с анеуплоидией доживет
до рождения.
Абсолютное большинство погибает на ранних сроках беременности.
Нарушения развития всегда затрагивают многие органы и ткани.
Механизм возникновения геномных мутаций связан с патологией нарушения
нормального расхождения хромосом в мейозе, в результате чего образуются
аномальные гаметы (по количеству хромосом), после оплодотворения которых
возникают гетероплоидные зиготы.
Анеуплоидии могут быть как по аутосомам, так и по гетеросомам.
Полные трисомии описаны у человека по большому числу хромосом: 8, 9, 13, 14,
18, 21, X.
Аномалии крупных хромосом (1-12) обычно летальны.
Трисомии 13 и 18 пар хромосом сублетальны.
Достаточная жизнеспособность имеет место только при трисомии по 21, аномалиях
половых хромосом и частичных аутосомных трисомиях.
95.
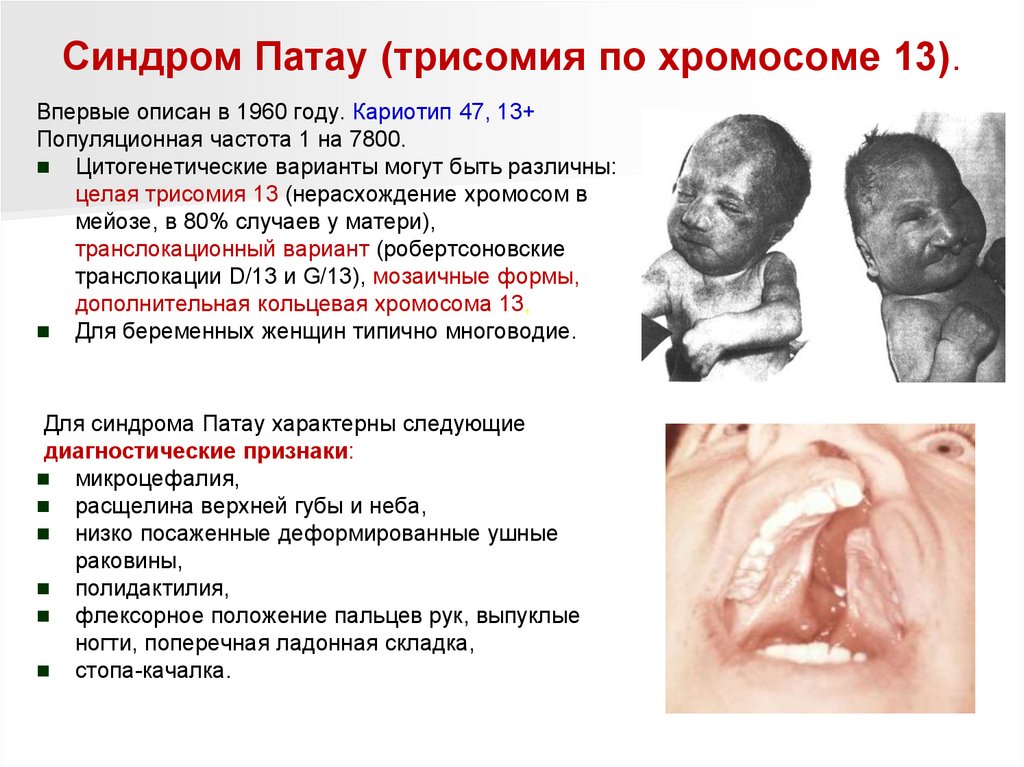
96.
Синдром Патау (трисомия по хромосоме 13).Впервые описан в 1960 году. Кариотип 47, 13+
Популяционная частота 1 на 7800.
Цитогенетические варианты могут быть различны:
целая трисомия 13 (нерасхождение хромосом в
мейозе, в 80% случаев у матери),
транслокационный вариант (робертсоновские
транслокации D/13 и G/13), мозаичные формы,
дополнительная кольцевая хромосома 13,
Для беременных женщин типично многоводие.
Для синдрома Патау характерны следующие
диагностические признаки:
микроцефалия,
расщелина верхней губы и неба,
низко посаженные деформированные ушные
раковины,
полидактилия,
флексорное положение пальцев рук, выпуклые
ногти, поперечная ладонная складка,
стопа-качалка.
97.
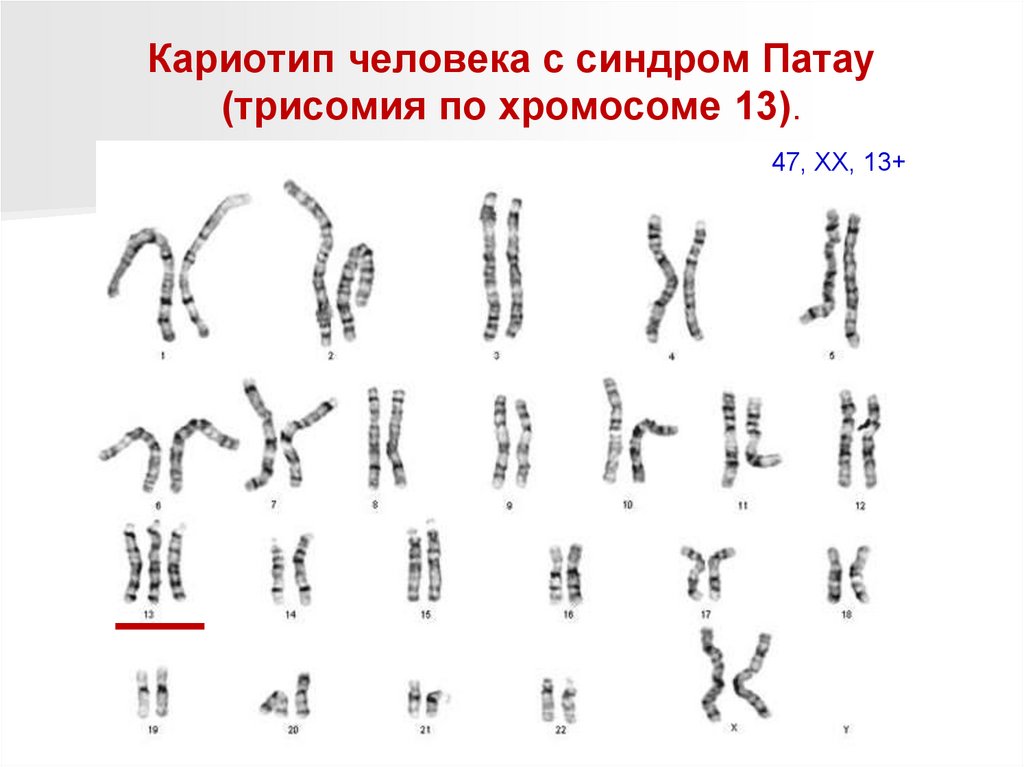
Кариотип человека с синдром Патау(трисомия по хромосоме 13).
47, ХХ, 13+
98.
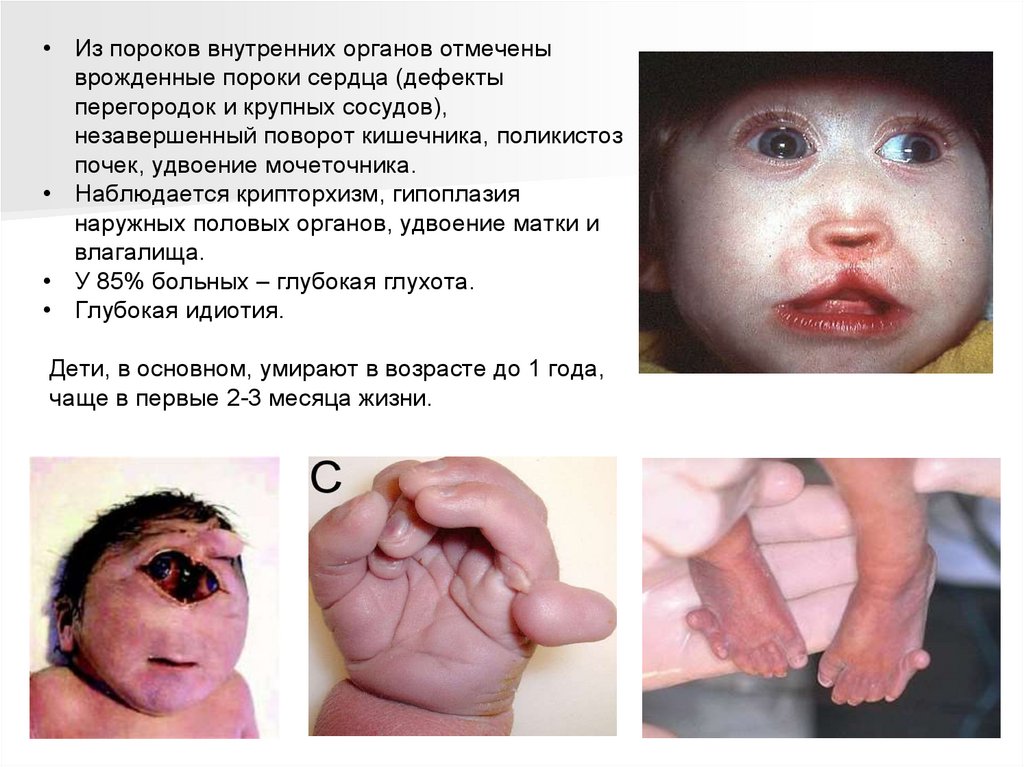
• Из пороков внутренних органов отмеченыврожденные пороки сердца (дефекты
перегородок и крупных сосудов),
незавершенный поворот кишечника, поликистоз
почек, удвоение мочеточника.
• Наблюдается крипторхизм, гипоплазия
наружных половых органов, удвоение матки и
влагалища.
• У 85% больных – глубокая глухота.
• Глубокая идиотия.
Дети, в основном, умирают в возрасте до 1 года,
чаще в первые 2-3 месяца жизни.
99.
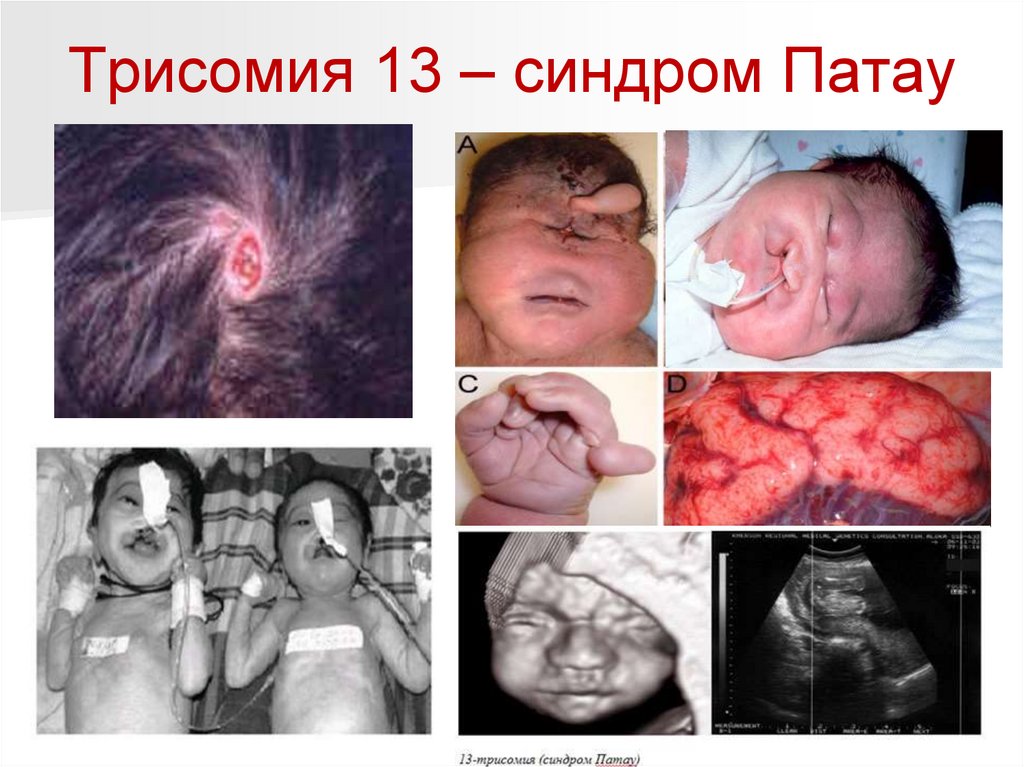
Трисомия 13 – синдром Патау100.
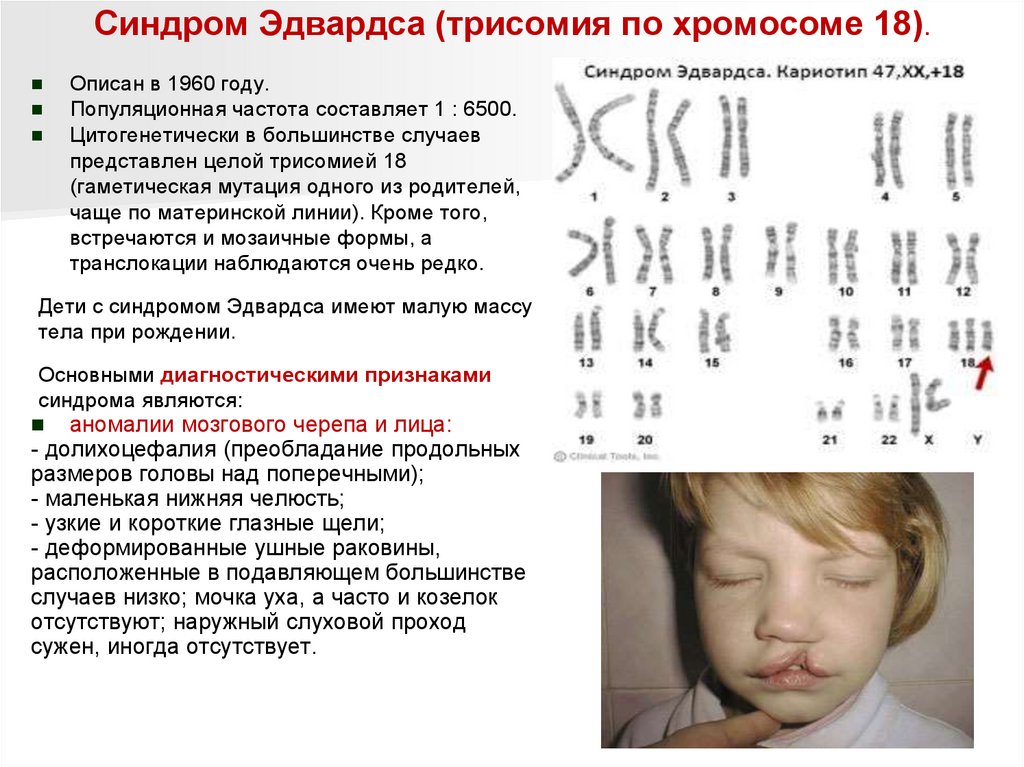
Синдром Эдвардса (трисомия по хромосоме 18).Описан в 1960 году.
Популяционная частота составляет 1 : 6500.
Цитогенетически в большинстве случаев
представлен целой трисомией 18
(гаметическая мутация одного из родителей,
чаще по материнской линии). Кроме того,
встречаются и мозаичные формы, а
транслокации наблюдаются очень редко.
Дети с синдромом Эдвардса имеют малую массу
тела при рождении.
Основными диагностическими признаками
синдрома являются:
аномалии мозгового черепа и лица:
- долихоцефалия (преобладание продольных
размеров головы над поперечными);
- маленькая нижняя челюсть;
- узкие и короткие глазные щели;
- деформированные ушные раковины,
расположенные в подавляющем большинстве
случаев низко; мочка уха, а часто и козелок
отсутствуют; наружный слуховой проход
сужен, иногда отсутствует.
101.
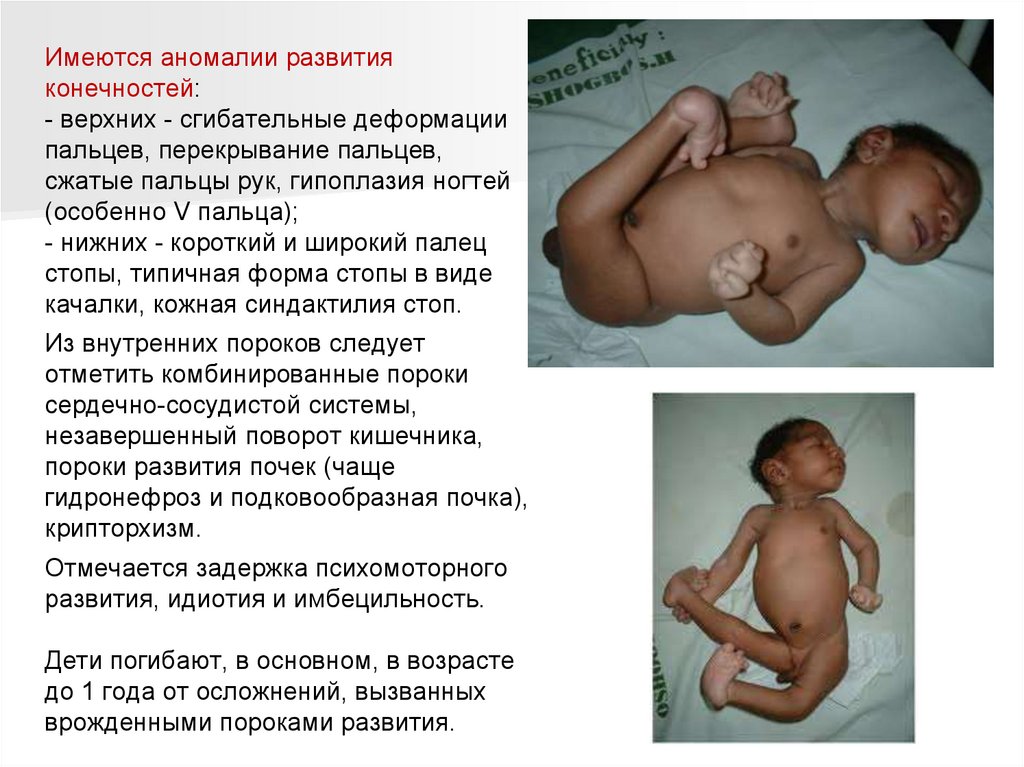
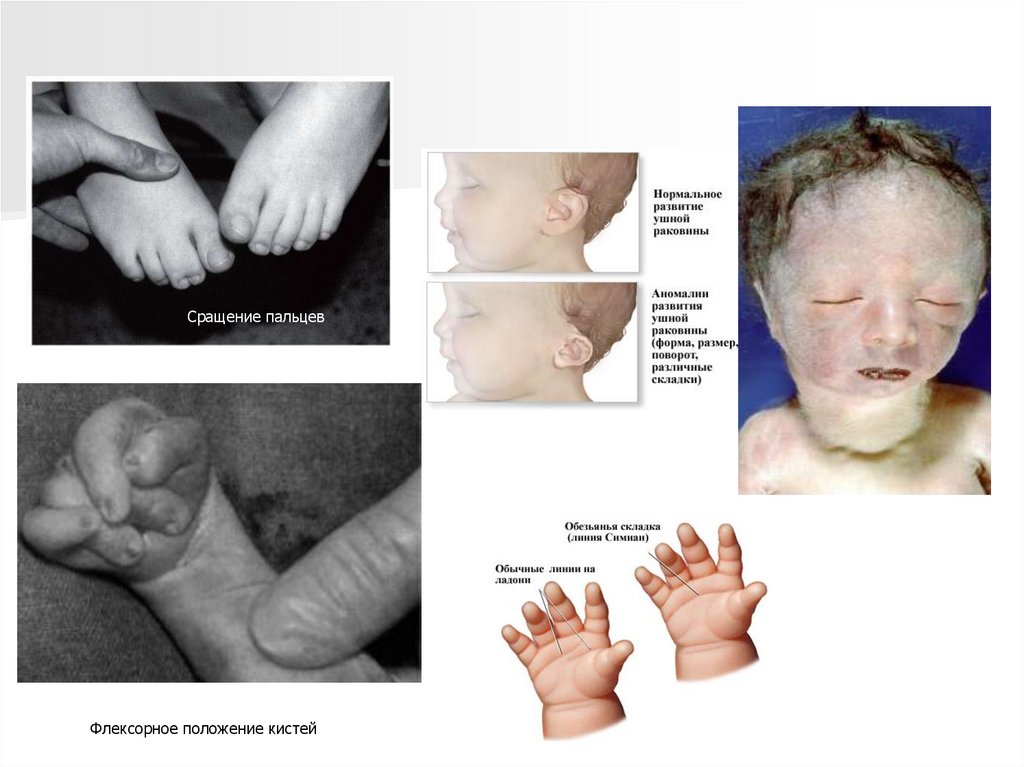
Имеются аномалии развитияконечностей:
- верхних - сгибательные деформации
пальцев, перекрывание пальцев,
сжатые пальцы рук, гипоплазия ногтей
(особенно V пальца);
- нижних - короткий и широкий палец
стопы, типичная форма стопы в виде
качалки, кожная синдактилия стоп.
Из внутренних пороков следует
отметить комбинированные пороки
сердечно-сосудистой системы,
незавершенный поворот кишечника,
пороки развития почек (чаще
гидронефроз и подковообразная почка),
крипторхизм.
Отмечается задержка психомоторного
развития, идиотия и имбецильность.
Дети погибают, в основном, в возрасте
до 1 года от осложнений, вызванных
врожденными пороками развития.
102.
Сращение пальцевФлексорное положение кистей
103.
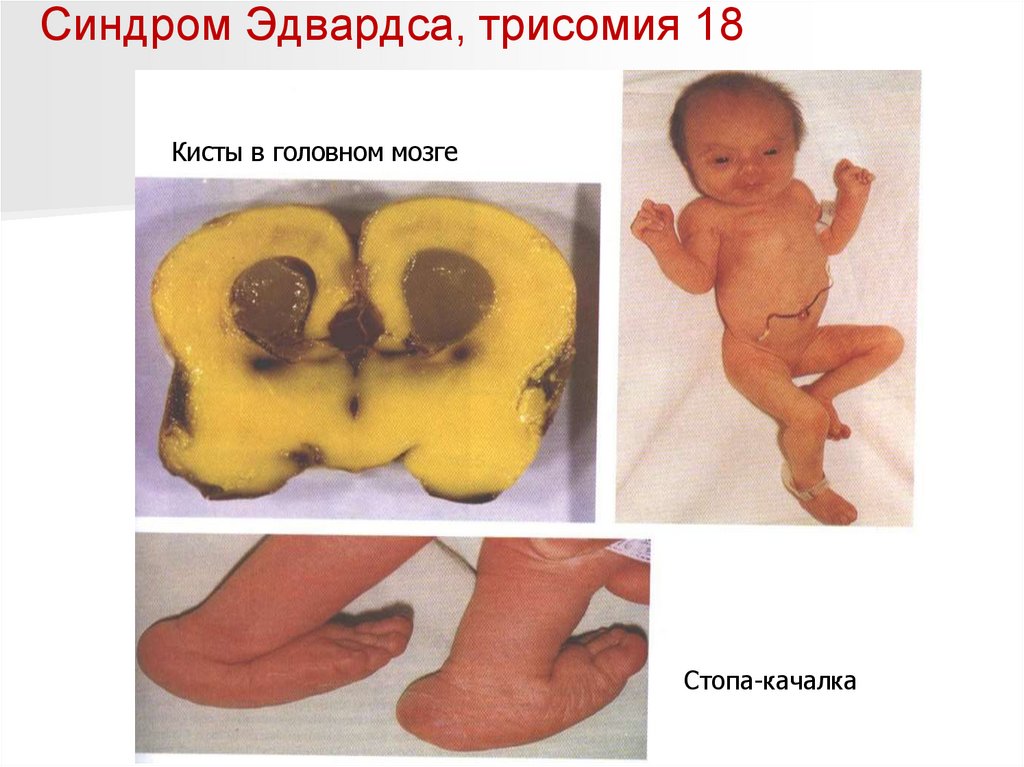
Синдром Эдвардса, трисомия 18Кисты в головном мозге
Стопа-качалка
104.
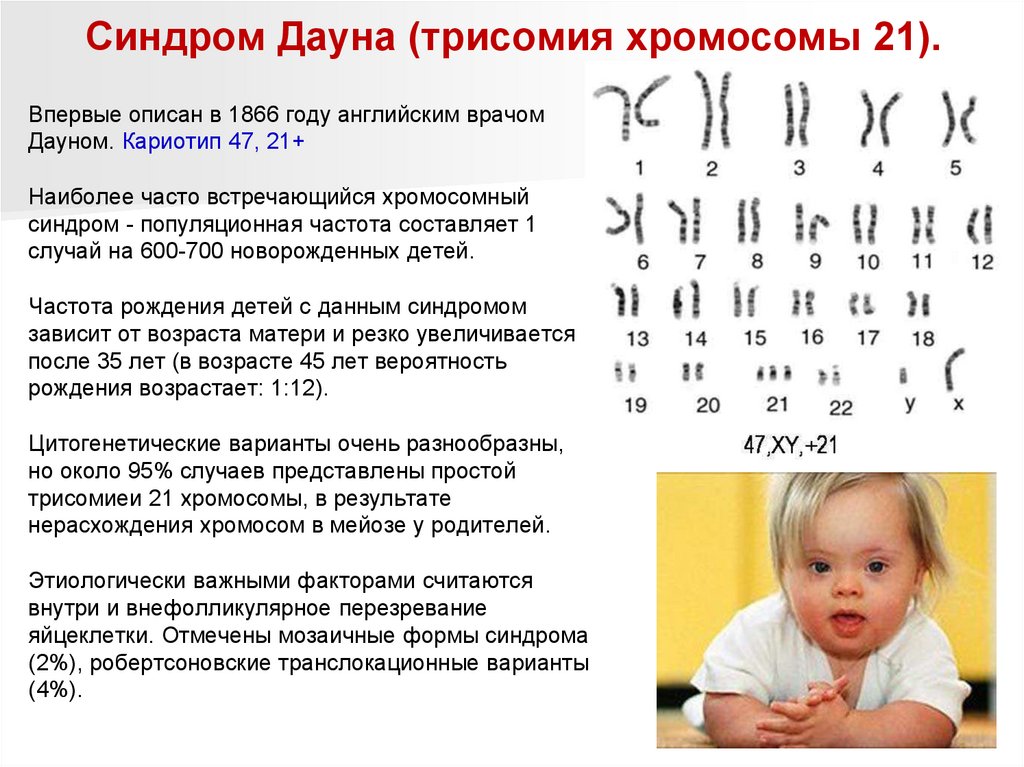
Синдром Дауна (трисомия хромосомы 21).Впервые описан в 1866 году английским врачом
Дауном. Кариотип 47, 21+
Наиболее часто встречающийся хромосомный
синдром - популяционная частота составляет 1
случай на 600-700 новорожденных детей.
Частота рождения детей с данным синдромом
зависит от возраста матери и резко увеличивается
после 35 лет (в возрасте 45 лет вероятность
рождения возрастает: 1:12).
Цитогенетические варианты очень разнообразны,
но около 95% случаев представлены простой
трисомиеи 21 хромосомы, в результате
нерасхождения хромосом в мейозе у родителей.
Этиологически важными факторами считаются
внутри и внефолликулярное перезревание
яйцеклетки. Отмечены мозаичные формы синдрома
(2%), робертсоновские транслокационные варианты
(4%).
105.
Основными диагностическими признаками синдромаявляются:
- уплощение профиля лица (90%),
- монголоидный разрез глаз (80%)
- эпикант,
- открытый рот,
- аномалии зубов,
- короткий нос и плоская переносица,
- деформированные уши (80%)
- избыток кожи на шее (80%)
- короткие конечности,
- поперечная четырех-пальцевая ладонная
складка (обезьянья борозда (45%).
- разболтанность суставов (80%)
- рост больных на 20 см ниже среднего.
Из пороков внутренних органов часто отмечаются
врожденные пороки сердца и желудочнокишечного тракта, которые и определяют
продолжительность жизни больных.
Умственная отсталость обычно средней степени
тяжести. Дети с синдромом Дауна часто ласковые
и привязчивые, послушные и внимательные.
Лечение должно быть многоплановым и неспецифичным. Многие люди с трисомией-21
способны вести самостоятельную жизнь, овладевать несложными профессиями,
создавать семьи, хотя их браки бесплодны.
106.
Синдром Дауна- трисомия 21107.
• Большое значение для диагностики имеет динамикафизического и умственного развития ребёнка. При
синдроме Дауна и то и другое задерживается. Рост
взрослых больных на 20 см ниже среднего. Задержка в
умственном развитии достигает имбецильности, если не
применяются специальные методы обучения. Дети с
синдромом Дауна ласковые, внимательные, послушные,
терпеливые при обучении. Коэффициент умственного
развития (IQ) у разных детей широко варьирует (от 20 до 75).
• Реакция детей с синдромом Дауна на факторы окружающей
среды часто патологическая в связи со слабым клеточным и
гуморальным иммунитетом, снижением репарации ДНК,
недостаточной выработкой пищеварительных ферментов,
ограниченными компенсаторными возможностями всех
систем. По этой причине дети с синдромом Дауна часто болеют
пневмониями, тяжело переносят детские инфекции. У них
отмечается недостаток массы тела, выражен авитаминоз.
Врождённые пороки внутренних органов, сниженная
приспособленность детей с синдромом Дауна часто приводят к
летальному исходу в первые 5 лет. Следствием изменённого
иммунитета и недостаточности репарационных систем (для
повреждённой ДНК) являются лейкозы, часто встречающиеся у
больных с синдромом Дауна. Средняя продолжительность жизни
при синдроме Дауна 36 лет.
108.
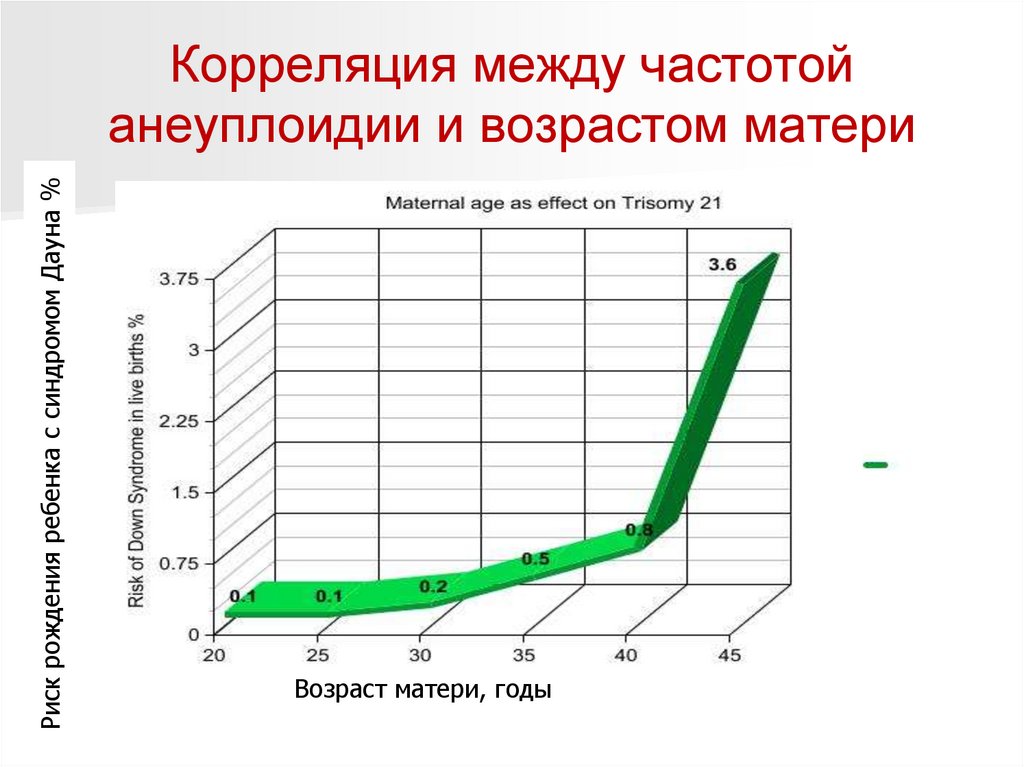
Факторы и патологии, которые могут привести ксиндрому Дауна
Браки между близкими родственниками. Близкие родственники являются
носителями одних и тех же генетических патологий. Поэтому если у двух
людей были дефекты 21-й хромосомы или белка, отвечающего за
распределение хромосом, то у их ребенка высока вероятность синдрома
Дауна. Причем, чем ближе степень родства, тем выше риск развития
генетической патологии.
Ранние беременности младше 18 лет.
У молодых девушек организм еще не до конца сформировался. Половые
железы могут работать не стабильно. Процессы созревания яйцеклеток часто
дают сбой, что может привести к генетическим аномалиям у ребенка.
Возраст матери старше 35 лет.
На протяжении жизни на яйцеклетки воздействуют различные вредные
факторы. Они негативно влияют на генетический материал и могут нарушить
процесс деления хромосом. Поэтому после 35 лет будущей маме необходимо
пройти медико-генетическое консультирование, чтобы до родов определить
генетические патологии у ребенка. Чем старше женщина, тем выше риск для
здоровья ее потомства.
109.
Риск рождения ребенка с синдромом Дауна %Корреляция между частотой
анеуплоидии и возрастом матери
Возраст матери, годы
110.
Факторы и патологии, которые могут привести ксиндрому Дауна
Возраст отца старше 45 лет.
С возрастом у мужчин нарушается процесс образования сперматозоидов и повышается
вероятность нарушений в генетическом материале. Если мужчина в этом возрасте
решился стать отцом, то желательно предварительно сделать анализ для определения
качества спермы и пройти курс витаминотерапии: 30 дней приема витамина Е и
минералов.
Возраст бабушки по материнской линии, на момент когда она родила ребенка.
Чем старше была бабушка, когда она была беременна, тем выше риск для ее внучек.
Дело в том, что все яйцеклетки матери сформировались в период внутриутробного
развития. Еще до рождения женщины у нее уже заложен запас яйцеклеток на всю
жизнь. Поэтому если возраст бабушки превышал 35 лет, то высок риск того, что именно
у матери больного малыша будет яйцеклетка с неправильным набором хромосом.
Родители являются носителями транслокации 21-й хромосомы.
Этот термин означает, что у одного из родителей участок 21-й хромосомы
прикрепляется к другой хромосоме, чаще всего к 14-й. Такая особенность никак не
проявляется внешне и человек не знает о ней. Но у таких родителей значительно
повышается риск рождения ребенка с синдромом Дауна. Это явление называется
«семейный синдром Дауна». Его доля среди всех случаев болезни не превышает 2%.
Но все молодые пары, у которых родился ребенок с синдромом, обследуют на наличие
транслокаций. Это помогает определить риск развития генетических отклонений при
следующих беременностях.
Синдром Дауна считается случайной генетической мутацией.
111.
Диагностика СДУЗИ-диагностика:
Сроки: первый триместр, оптимально с 11-й по 13-ю неделю
беременности.
Повторные УЗИ делают на 24-й и 34-й неделе беременности. Но эти
исследования считаются менее информативными для диагностики
синдрома Дауна.
Показания: всем беременным женщинам.
Противопоказания: пиодермия (гнойное поражение кожи).
112.
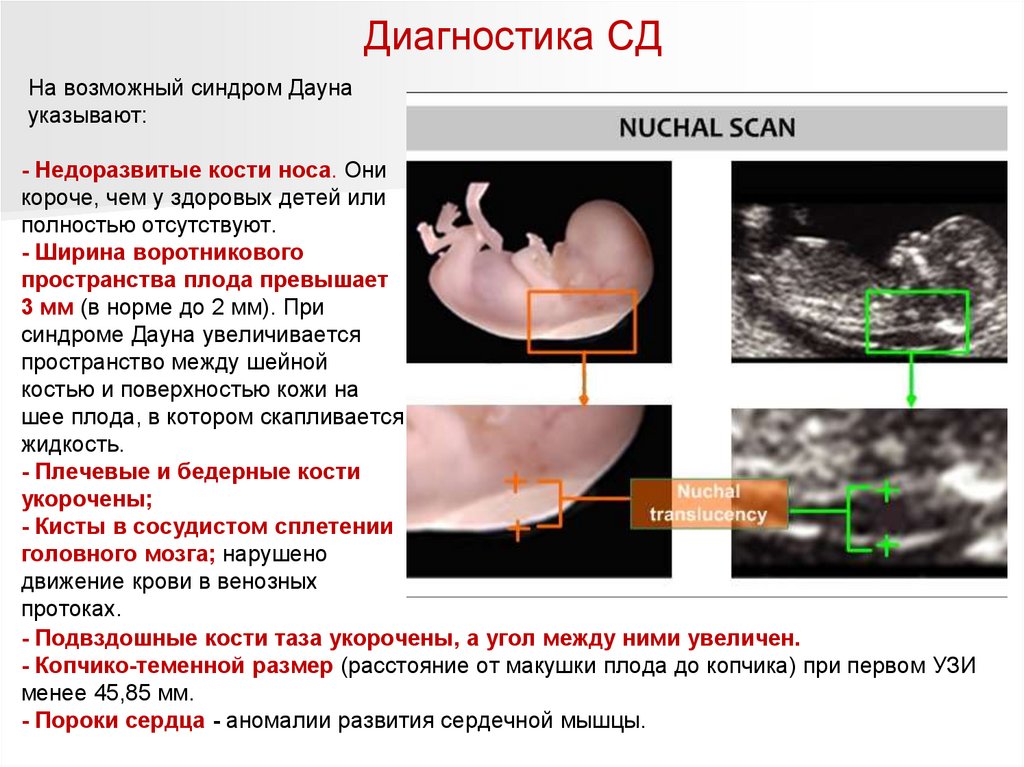
Диагностика СДНа возможный синдром Дауна
указывают:
- Недоразвитые кости носа. Они
короче, чем у здоровых детей или
полностью отсутствуют.
- Ширина воротникового
пространства плода превышает
3 мм (в норме до 2 мм). При
синдроме Дауна увеличивается
пространство между шейной
костью и поверхностью кожи на
шее плода, в котором скапливается
жидкость.
- Плечевые и бедерные кости
укорочены;
- Кисты в сосудистом сплетении
головного мозга; нарушено
движение крови в венозных
протоках.
- Подвздошные кости таза укорочены, а угол между ними увеличен.
- Копчико-теменной размер (расстояние от макушки плода до копчика) при первом УЗИ
менее 45,85 мм.
- Пороки сердца - аномалии развития сердечной мышцы.
113.
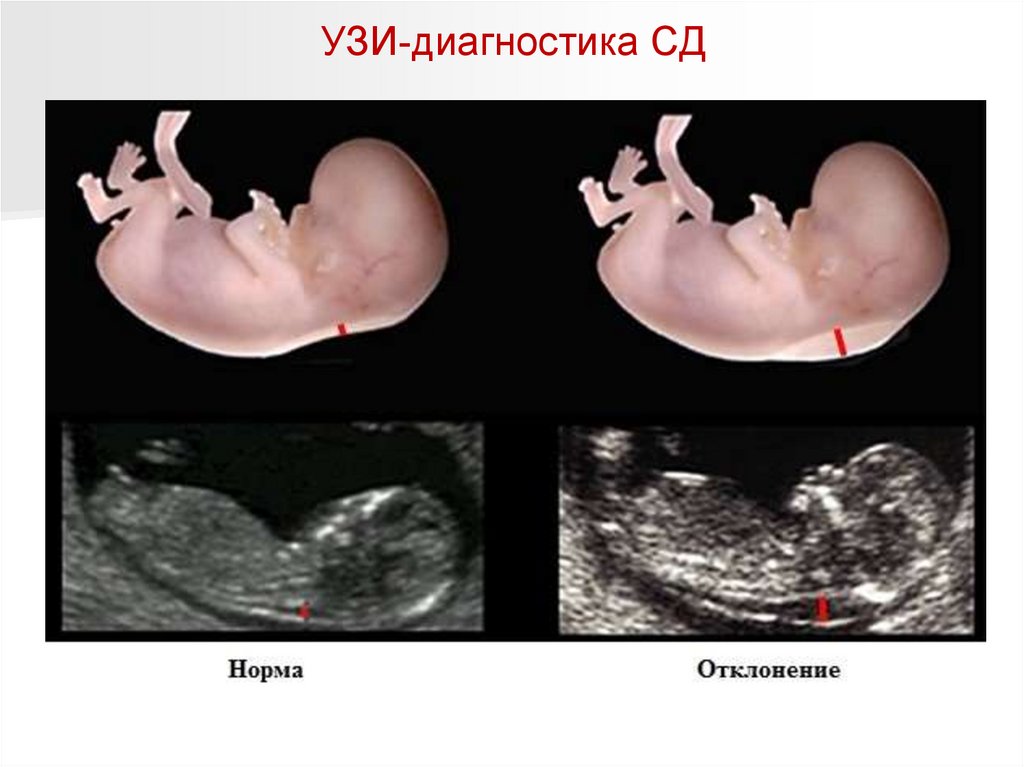
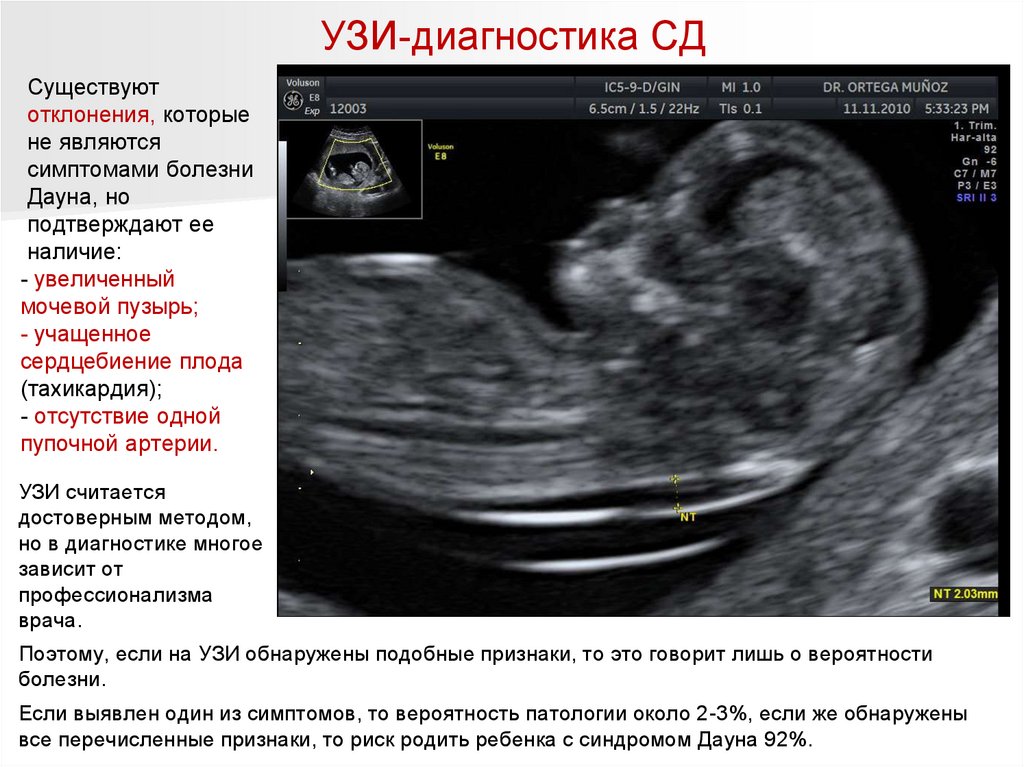
УЗИ-диагностика СД114.
УЗИ-диагностика СДСуществуют
отклонения, которые
не являются
симптомами болезни
Дауна, но
подтверждают ее
наличие:
- увеличенный
мочевой пузырь;
- учащенное
сердцебиение плода
(тахикардия);
- отсутствие одной
пупочной артерии.
УЗИ считается
достоверным методом,
но в диагностике многое
зависит от
профессионализма
врача.
Поэтому, если на УЗИ обнаружены подобные признаки, то это говорит лишь о вероятности
болезни.
Если выявлен один из симптомов, то вероятность патологии около 2-3%, если же обнаружены
все перечисленные признаки, то риск родить ребенка с синдромом Дауна 92%.
115.
116.
Биохимический анализ кровиНа первом и втором триместре у матери берут кровь для
биохимического исследования. В ней определяют:
Хорионический гонадотропин человека (ХГЧ) гормон, который
выделяется плацентой в организме беременной женщины.
Ассоциированный с беременностью белок А (РАРР-А). Этот белок
вырабатывается плацентой на начальных этапах беременности для
подавления атаки материнского иммунитета по отношению к плоду.
Свободный эстриол – женский стероидный гормон, вырабатываемый в
плаценте из гормона-предшественника который выделяют надпочечники
плода.
Альфа-фетопротеин (АФП) – белок, вырабатываемый в печени и
пищеварительной системе плода для защиты от иммунитета матери.
Сроки:
Первый триместр с 10-й по 13-ю неделю беременности. Исследуют
сыворотку крови на ХГЧ и РАРР-А. Это так называемый двойной тест.
Он считается более точным, чем исследование крови на втором
триместре. Его достоверность 85%.
Второй триместр с 16-й по 18-ю неделю беременности. Определяют
уровень ХГЧ, АФП и свободный эстриол. Это исследование получило
название тройной тест. Достоверность 65%.
117.
Показания. Это обследование не является обязательным, но его рекомендуют пройтив таких случаях:
возраст матери старше 30 лет;
в семье есть дети с синдромом Дауна;
есть близкие родственники с генетическими болезнями;
перенесенное во время беременности тяжелое заболевание;
предыдущие беременности (2 и более) заканчивались выкидышами.
Противопоказания: не существует.
Интерпретация результатов.
Результаты двойного теста на первом триместре:
ХГЧ значительное превышение нормы (свыше 288,000 мЕд/мл) говорит о
генетической патологии, многоплодной беременности, неправильно поставленном
сроке беременности.
РАРР-А снижение меньше 0,6 МоМ говорит о синдроме Дауна, угрозе выкидыша
или неразвивающейся беременности.
Результаты тройного теста на втором триместре:
ХГЧ более 2 МоМ свидетельствует о риске синдрома Дауна и синдрома
Клайнфельтера;
АФП менее 0,5 МоМ может говорить о наличии у ребенка синдрома Дауна или
синдрома Эдвардса.
Свободный эстриол менее 0,5 МоМ свидетельствует, что надпочечники у плода
работают недостаточно, что бывает при синдроме Дауна.
118.
Особенности социализации людей с СДДля успешной социализации ребенок с синдромом
Дауна должен посещать обычную государственную
школу. Интеграция в обычную школу даст ему
возможность учиться жить и действовать так, как это
принято в окружающем его мире. А для этого
необходимо, чтобы ребенок-даун посещал
специализированное ДОУ:
1) сформировать у людей с синдромом Дауна
необходимые социальные навыки и навыки
самообслуживания, необходимые для работы на
рабочем месте;
2) адаптировать социум для включения в него
людей с синдромом Дауна
119.
Театр простодушных (г. Москва)120.
Клинико-цитогенетическаяхарактеристика синдромов,
связанных со структурными
перестройками хромосом
121.
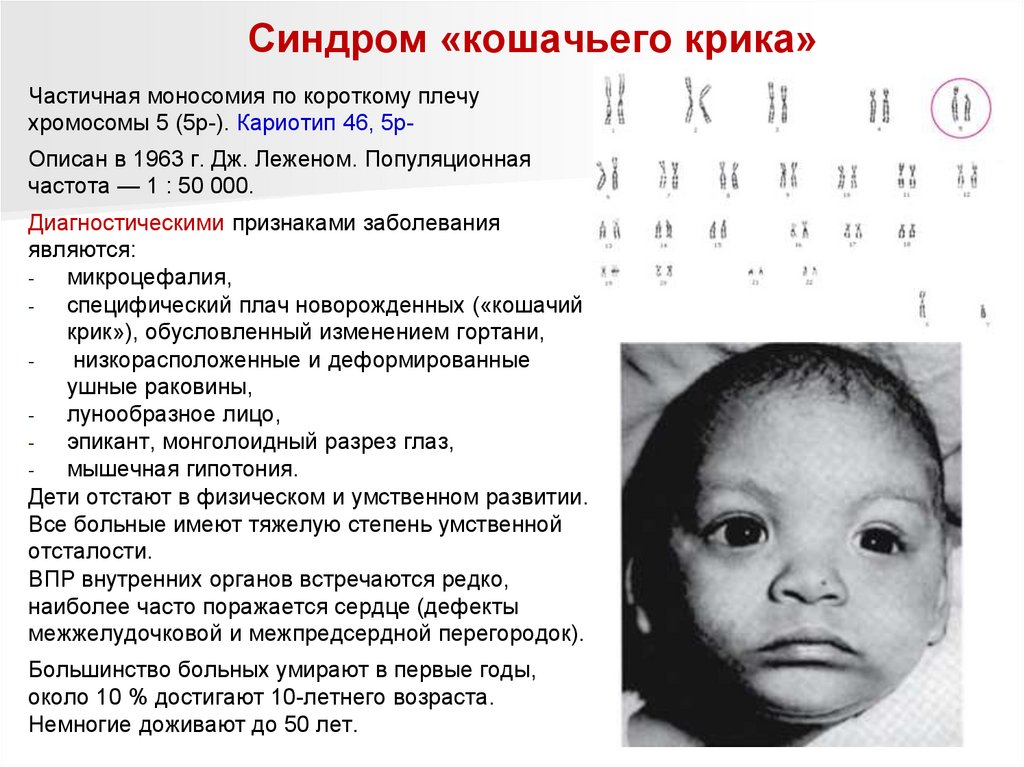
Синдром «кошачьего крика»Частичная моносомия по короткому плечу
хромосомы 5 (5р-). Кариотип 46, 5рОписан в 1963 г. Дж. Леженом. Популяционная
частота — 1 : 50 000.
Диагностическими признаками заболевания
являются:
микроцефалия,
специфический плач новорожденных («кошачий
крик»), обусловленный изменением гортани,
низкорасположенные и деформированные
ушные раковины,
лунообразное лицо,
эпикант, монголоидный разрез глаз,
мышечная гипотония.
Дети отстают в физическом и умственном развитии.
Все больные имеют тяжелую степень умственной
отсталости.
ВПР внутренних органов встречаются редко,
наиболее часто поражается сердце (дефекты
межжелудочковой и межпредсердной перегородок).
Большинство больных умирают в первые годы,
около 10 % достигают 10-летнего возраста.
Немногие доживают до 50 лет.
122.
123.
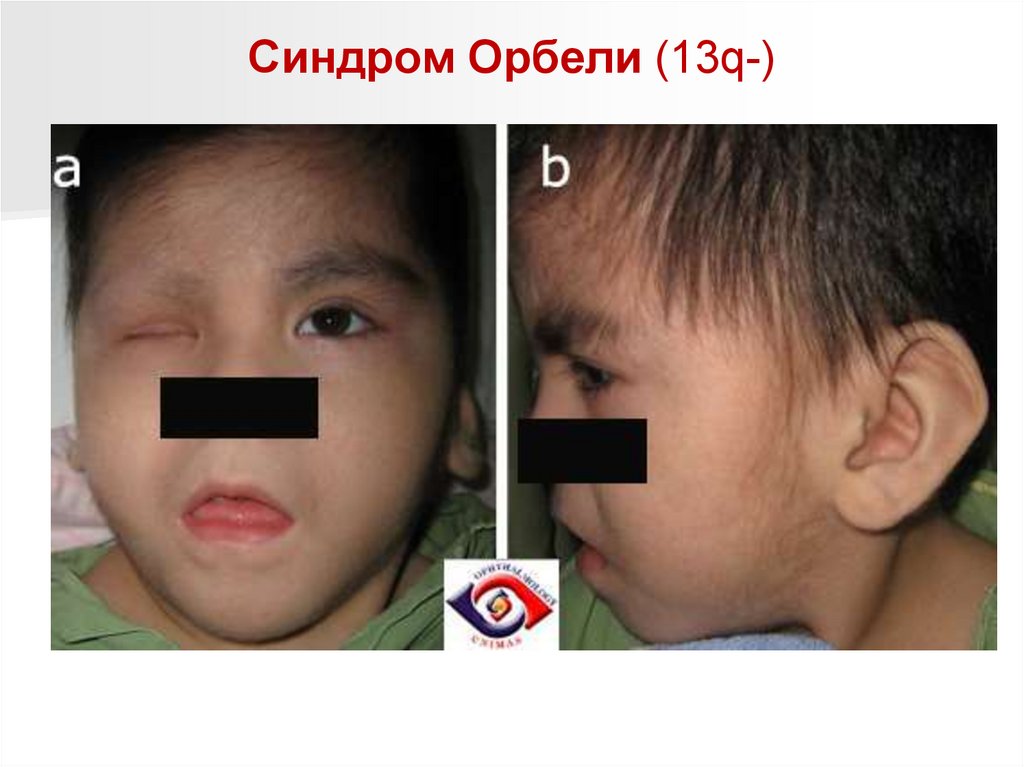
Синдром Орбели (13q-)Обусловлен делецией длинного плеча тринадцатой хромосомы.
Популяционная частота синдрома не установлена.
Дети с синдромом Орбели рождаются с низким (2200 г) весом.
Клинически синдром проявляется аномалиями развития всех систем
организма.
Характерны:
- микроцефалия,
- отсутствие носовой вырезки (лоб непосредственно переходит в нос),
широкая спинка носа, эпикант, антимонголоидный разрез глаз,
- высокое нёбо,
- низко расположенные деформированные ушные раковины.
Отмечаются поражения глаз, опорно-двигательного аппарата (короткая
шея, гипо- или аплазия первого пальца кисти и пяточной кости,
синдактилии кистей и стоп), атрезии прямой кишки и заднепроходного
отверстия.
Часты пороки развития сердца, почек, головного мозга.
Для всех детей с синдромом Орбели характерна глубокая
олигофрения, возможны потери сознания и судороги.
Большинство больных с синдромом 13q- погибают на 1-м году жизни.
124.
Синдром Орбели (13q-)125.
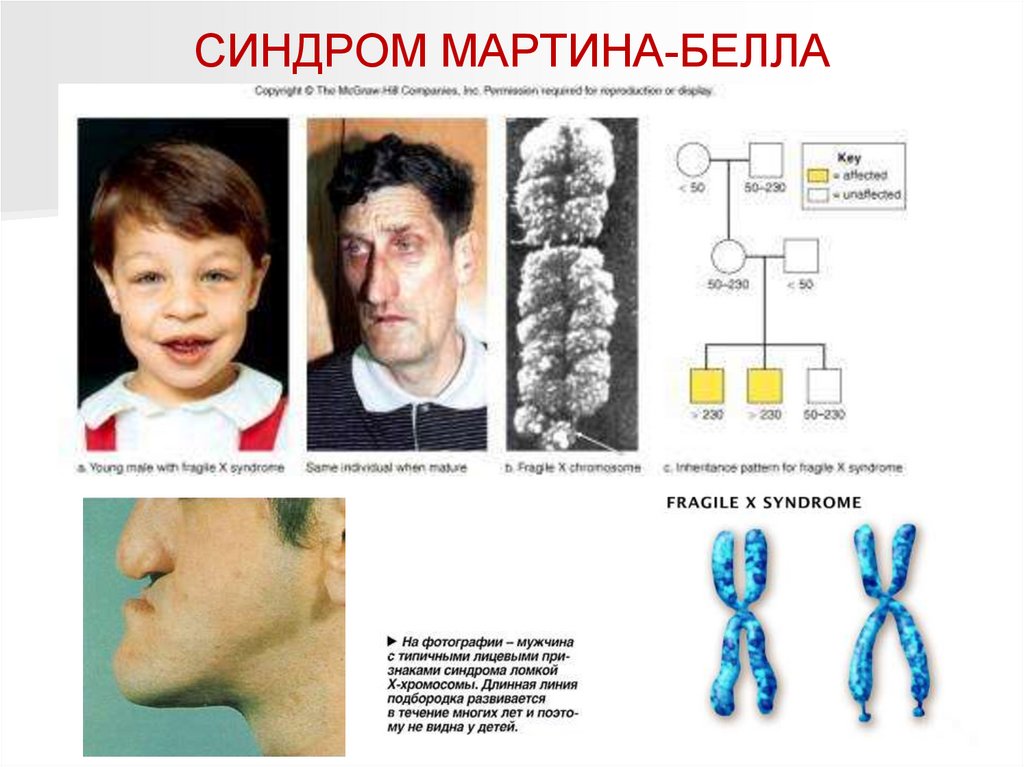
СИНДРОМ МАРТИНА-БЕЛЛА (синдром ломкой,или фрагильной Х-хромосомы)
Синдром Мартина-Белла – самая
распространенная (после болезни Дауна)
форма умственной отсталости. Мальчики
болеют в 2-3 раза чаще девочек.
Клинические признаки: удлиненное лицо,
высокий выступающий лоб, выступающий
подбородок, оттопыренные крупные уши,
крупные кисти и стопы, макроорхидизм,
пролапс митрального клапана,
плоскостопие, глубокая или умеренная
олигофрения (Умственное развитие
Лицо больного с синдромом
Мартина-Белла
колеблется между значениями IQ от 30 до
65 (иногда в границах нормы).
Цитогенетическая картина: ломкость
дистального конца длинного плечика Ххромосомы (Хq – напоминает спутник).
Ломкая Х-хромосома (слева – женская,
справа – мужская) при синдроме
Мартина-Белла
Тип наследования: Х-сцепленный
Популяционная частота – 1 : 1250
(мальчики); 1 : 2500-3000 (девочки)
126.
СИНДРОМ МАРТИНА-БЕЛЛА127.
Речь изобилует повторами, часто встречается своеобразное заикание(логоневроз).
Для детей характерна двигательная расторможенность и некоторые
симптомы аутизма (ребенок избегает глазного контакта, производит
стереотипные движения руками, испытывает страхи).
Даже при легкой степени интеллектуальной недостаточности дети с
трудом овладевают навыками счета и письма. Дети с ломкой Ххромосомой имеют своеобразную электроэнцефалограмму.
В связи с тем, что симптомы заболевания разнообразны, часто
ставится ошибочный диагноз (шизофрения, ранний детский аутизм,
эпилепсия, синдром дефицита внимания и гиперактивности).
В результате дети не получают соответствующего
лечения, а семья остается в неведении относительно
истинных причин нарушения развития.
128.
Подходы в борьбе с наследственнымиболезнями
1. Массовое «просеивание» новорожденных на
наследственные дефекты обмена веществ (выявление
фенилкетонурии, гипотиреоза, муковисцидоза,
галактоземии и др.).
2. Пренатальная диагностика (с использованием
разных методов: УЗИ, фетоскопия, амниоцентез и др.)
в 1- и 2-м триместрах беременности.
3. Медико-генетическое консультирование.
4. Контроль за мутагенной опасностью факторов
окружающей среды.