


























![[(A1>A2)=B] > O și D>d și M=N și Xg+>Xg-](https://cf4.ppt-online.org/files4/slide/b/bXAhBUJoCurx8M3VdjRIiymfQP0LGF16tDK2vg/slide-32.jpg "[(A1>A2)=B] > O și D>d și M=N și Xg+>Xg-")


























")









")


 при мутациях в одном и том же гене")
















 biology
biologySimilar presentations:






")



Наследственные нормальные и патологические признаки человека
1. Лекция 9 Наследственные нормальные и патологические признаки человека
Catedra de biologie moleculară și genetica umanăЛекция 9
Наследственные нормальные
и патологические признаки
человека
План лекции
1. Корреляция генотип-фенотип
- взаимодействия аллельных генов
- взаимодействия неаллельных генов
2. Моногенные менделирующие признаки
3. Моногенные неменделирующие признаки
4. Генетические феномены в проявлении и передаче
наследственных признаков
d.ș.m, lector universitar
Sidorenko Ludmila
2.
НАСЛЕДСТВЕННЫЕ ПРИЗНАКИОПРЕДЕЛЯЮТСЯ ЭКСПРЕССИЕЙ ОДНОГО ИЛИ НЕСКОЛЬКИХ
ГЕНОВ
!!! Роль генетических факторов более 50%.
Наследственные
признаки:
• Моногенные и полигенные;
• Монофаториальные и
мультифакториальные;
• Морфологические, физиологические,
биохимические и поведенческие.
3. Моногенные признаки vs полигенные признаки!
4.
Наследственные признакиМоногенные
Полигенные
• Определяются одной парой
аллельных генов
• Обычно
монофакториальные
• Часто наследуются по
законам Менделя
• Могут быть нормальными и
патологическими
• Могут быть доминантными,
кодоминантными,
рецессивными
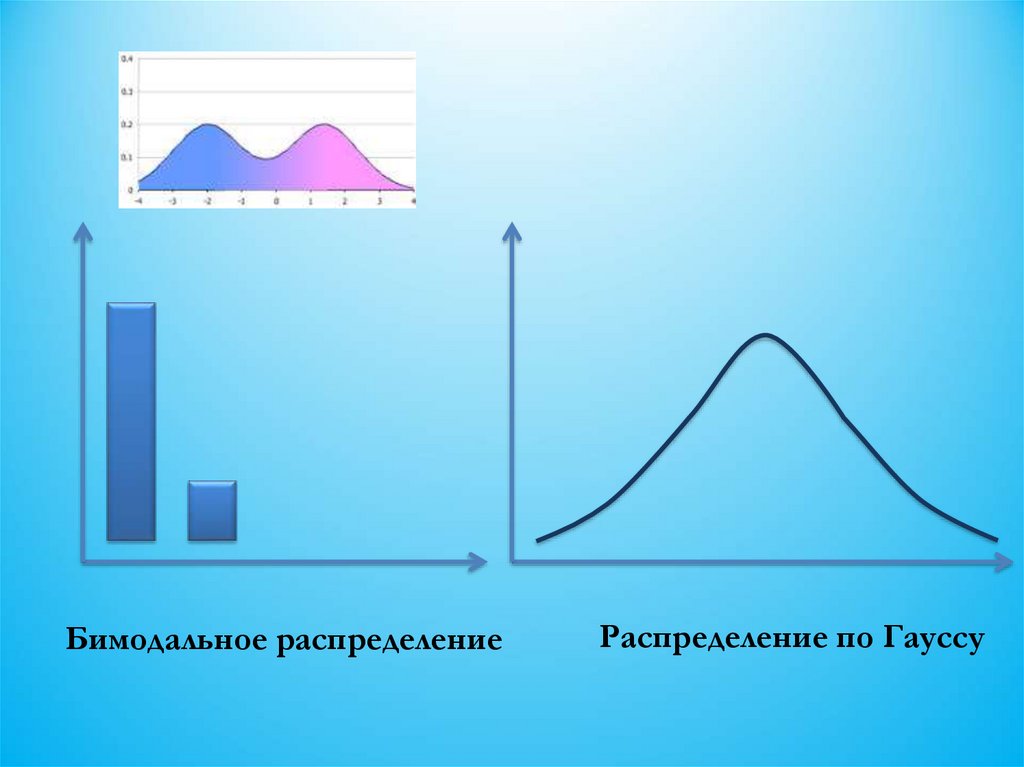
• Имеют бимодальное
распределение
• Определяются несколькими
парами неаллельных генов
• Обычно
мультифакториальные и
неменделирующие
• Могут быть нормальными и
патологическими
• Могут иметь разные
фенотипы
• Имеют гауссово
распределение
5.
Бимодальное распределениеРаспределение по Гауссу
6.
Примеры признаковМоногенные
• Rh+ / Rh-;
• ABO, MN, Xg ...
• Гаптоглобины
• Трансферины
• Секретор / несекретор
• полидактилия/ пендактилия
• Норм.фенотип/ ФК
• Норм.коагуляция / гемофилия
• С. Марфана и др. моногенные
заболевания из Вашей
таблицы по 18 генам
Полигенные
• Цвет кожи
• Рост
• Вес
• Артериальное давление
• IQ
• Отпечатки пальцев
• Сахарный диабет
• Шизофрения
•…
7.
Моногенные признаки1-ая часть
1. Группы крови
- Rh
- AB0 и Se
- MN
- Xg
2. Rh конфликт и ГБНД
3. Фенотип Bombay
8.
Наследование фактора RhD – Rh+
d – RhParents
DD x dd
Gametes D
F1
D – нормоморфный , доминантный
d – аморфный, рецессивный
Dd – 100%
d
P
Dd x dd
P
Dd x Dd
G
Dd
d
G
Dd Dd
F1
Dd – 50%
dd – 50%
F1
DD – 25%
Dd – 50%
dd – 25%
9.
Гемолитическая болезнь новорожденного (ГБНД)или Rh конфликт
1st pregnancy
Postpartum
Next
pregnancies
10. Риски для ГБНД
1. Риск 0 %Мать Rh+
Мать и отец Rh- (так что все дети Rh-)
2. Риск 50 %
Мать Rh- и отец Rh+ (Dd- гетерозиготный)
3. Риск 100 %
Мать Rh- и отец Rh+ (DD- гомозиготный)
11. Алгорифм для стратификации риска ГБНД?
1. Женщина Rh+/Rh-?2. Если женщина (мать) Rh+, то риск 0 %
3. Если она Rh - , то смотрим на её мужа (отца
ребёнка)?
Если он Rh - (риск 0 %)
Если он Rh+ (Dd-гетерозиготный*) 50 % риск
Если он Rh+ (DD-гомозиготный**) риск 100 %
*dd + Dd = Dd,Dd, dd, dd
**dd + DD = Dd,Dd,Dd,Dd
12. Rh- жена и Rh+ муж
• dd x Dd (муж гетерзиготный) = риск иметьдетей с резус конфликтом (ГБНД) 50 %
Dd
Dd
dd
dd
Дети резус +, ГБНД
Дети резус -, нет ГБНД
13. Rh- жена и Rh+ муж
• dd x DD (муж гомозиготный) = риск 100 %• Dd
• Dd
• Dd
• Dd
Дети резус +, ГБНД
14. Rh- жена и Rh- муж
dd x dd (муж гомозиготный) = риск иметьдетей с резус конфликтом (ГБНД) 0 %
• dd
• dd
• dd
• dd
Все дети резус - , ГБНД нет
15.
Когда происходит ГБНД?• Если у матери резус+, а у
плода резус• Если у матери резус-фактор,
а у плода Rh+, то уже в
первом ребенке
• Если мать резус-фактор, а
плод Резус+, и это второй
ребенок
• Если у матери резус+, а у
плода резус+
• Если у матери резус-фактор,
а у плода резус-
Если у матери резус+, а у ее мужа
резус-фактор, насколько высок
риск ГБНД для их детей?
0%
50 %
100 %
25 %
75 %
16.
Что происходит при ГБНБ(гемолитической болезни
новорожденных)?
Антитела матери атакуют AgD на
эритроциты ребенка
Антитела ребенка атакуют AgD на
эритроциты матери
AgD атаки матери Антитела ребенка
AgD атаки ребенка Антитела матери
17.
Если у матери резус+, а у еемужа резус-фактор, насколько
высок риск развития ГБНД
(гемолитической болезни
новорожденных) у их детей?
0%
50 %
100 %
25 %
75 %
Если у матери резус+, а у ее
ребенка резус-фактор,
насколько высок риск развития
ГБНД (гемолитической болезни
новорожденных) у этого
ребенка?
0%
50 %
100 %
25 %
75 %
18.
Если у матери резус-фактор, а уее ребенка резус-фактор,
насколько высок риск развития
ГБНД (гемолитической болезни
новорожденных) у этого
ребенка?
Если у матери резус+, а у ее
ребенка резус+, насколько
высок риск развития ГБНД
(гемолитической болезни
новорожденных) у этого
ребенка?
0%
50 %
100 %
25 %
75 %
0%
50 %
100 %
25 %
75 %
19.
Каков риск рождения ребенка с ГБНД у женщины, котораяродила ребенка с ГБНД, ее муж Rh+ гомозигота?
Что является причиной ГБНД
Как проявляется ГБНД на молекулярном уровне
20.
Каков риск развития ГБНД в семье, где обародителя страдали ГБНД и первый ребенок здоров?
Каков риск развития ГБНД в семье, где отец страдал
ГБНД, а мать не страдала и имеет резус-фактор +?
Женщина страдает от ГБНД. Каков риск того, что ее
дети будут страдать от той же болезни?
Женщина, которая не страдала, чей отец и старший
брат страдали от ГБНД, выходит замуж за rhмужчина Каков риск для этой пары иметь ребенка,
страдающего ГБНД?
Примечание. Словосочетание «страдал ГБНД» означает, что этот человек резус положительный, но
гетерозигота (Dd), а его мама резус отрицательная (dd)
21.
Каков риск развития HDNB в семье, где отец ипервый сын страдали HDNB?
Каков риск для резус-женщины, родившей резус- и
резус+ ребенка, иметь еще одного ребенка с ГБНД?
У женщины есть ребенок, который страдал от ГБНД.
Ее муж страдал такой же болезнью. Каков риск для
этой пары, если у этой пары будет еще один
ребенок, страдающий ГБНД?
Женщина, которая не страдала от ГБНД, хотя имела
старшего брата с этим заболеванием, замужем за
мужчиной, который страдал ГБНД. Каков риск для
этой пары, если у этой пары родится ребенок,
страдающий ГБНД?
22.
Каков риск для женщины, родившей ребенкас ГБНД и имеющей гомозиготного мужа с
резус+, иметь еще одного ребенка с тем же
заболеванием?
23.
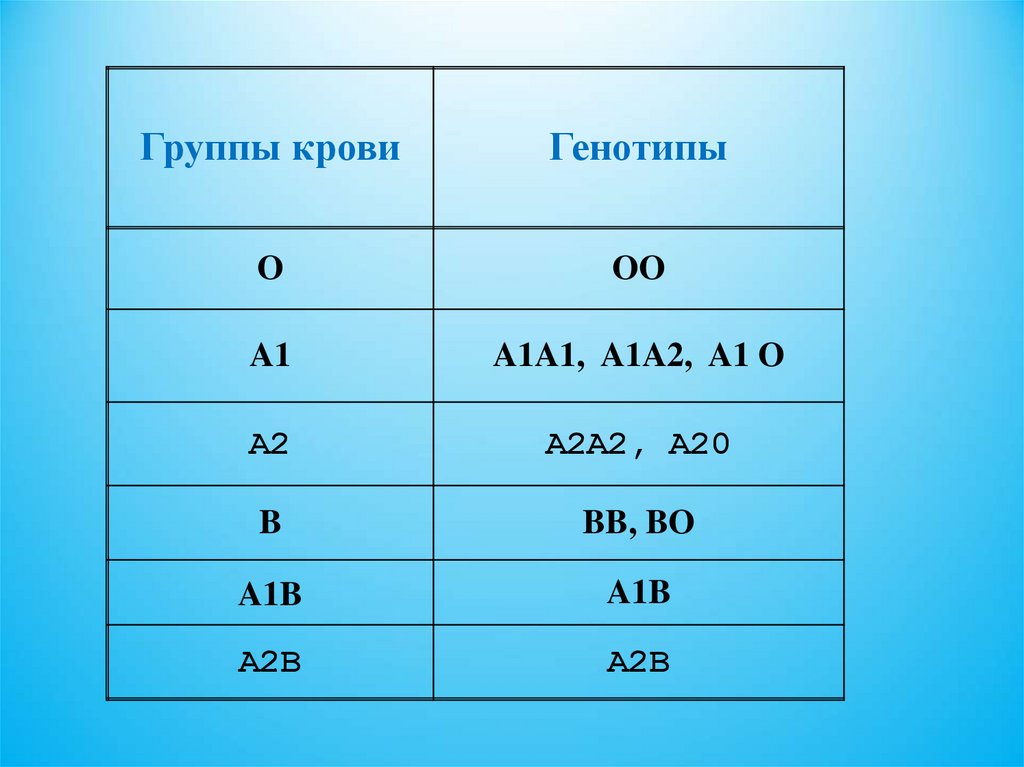
Система групп крови АВ024.
Группы кровиГенотипы
O
OO
A1
A1A1, A1A2, A1 O
A2
A2A2, A20
B
BB, BO
A1B
A1B
A2B
A2B
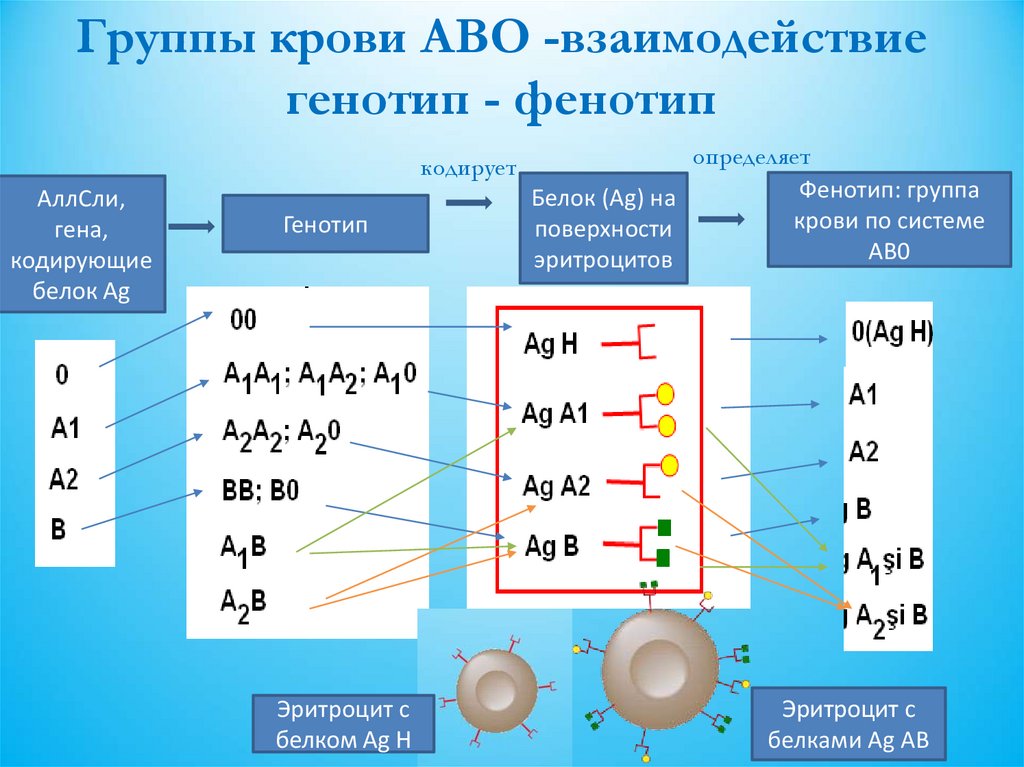
25.
Группы крови АВО -взаимодействиегенотип - фенотип
кодирует
АллСли,
гена,
кодирующие
белок Ag
Генотип
Эритроцит с
белком Ag Н
определяет
Фенотип: группа
Белок (Ag) на
крови по системе
поверхности
АВ0
эритроцитов
Эритроцит с
белками Ag АВ
26.
AB0 System27.
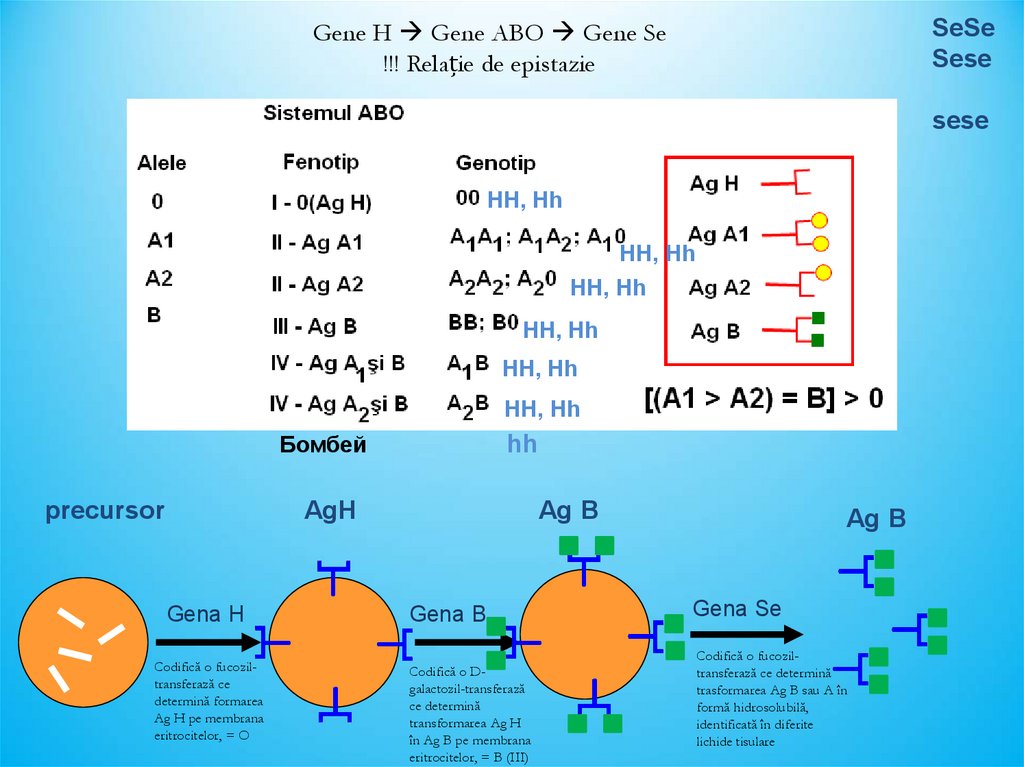
Gene H Gene ABO Gene Se!!! Relație de epistazie
SeSe
Sese
sese
HH, Hh
HH, Hh
HH, Hh
HH, Hh
HH, Hh
HH, Hh
Бомбей
precursor
hh
AgH
Gena H
Codifică o fucoziltransferază ce
determină formarea
Ag H pe membrana
eritrocitelor, = O
Ag B
Gena B
Codifică o Dgalactozil-transferază
ce determină
transformarea Ag H
în Ag B pe membrana
eritrocitelor, = B (III)
Ag B
Gena Se
Codifică o fucoziltransferază ce determină
trasformarea Ag B sau A în
formă hidrosolubilă,
identificată în diferite
lichide tisulare
28.
Нормальные моногенные признакиCaracter
Alele
Crs
Rh factor
D>d
1
DD, Dd
dd
Rh+
Rh-
19
SeSe, Sese
sese
Secretor
nesecretor
19
HH, Hh
hh
H ……..
Bombay fenotip
00
A1A1, A1A2, A10
A2A2, A20
BB, B0
0 (I)
A1 (II)
A2 (II)
B (III)
A1B
A2B
A1B (IV)
A2B (IV)
4
MM
MN
NN
M
MN
N
X
Xg(a+)Xg(a+),
Xg(a+)Xg(a-), Xg(a+)Y
Xg(a-)Xg(a-), Xg(a-)Y
Xg+
Xg-
Secretor
Antigen H
Dominare completă
Se>se
Dominare completă
H> h
Dominare completă
[(A1>A2)= B]>0
AB0
MN
Xg
Dominare completă
Codominare
M=N
Codominare
Xg(a+)>Xg(a-)
Dominare completă
9
Genotip
Fenotip
29.
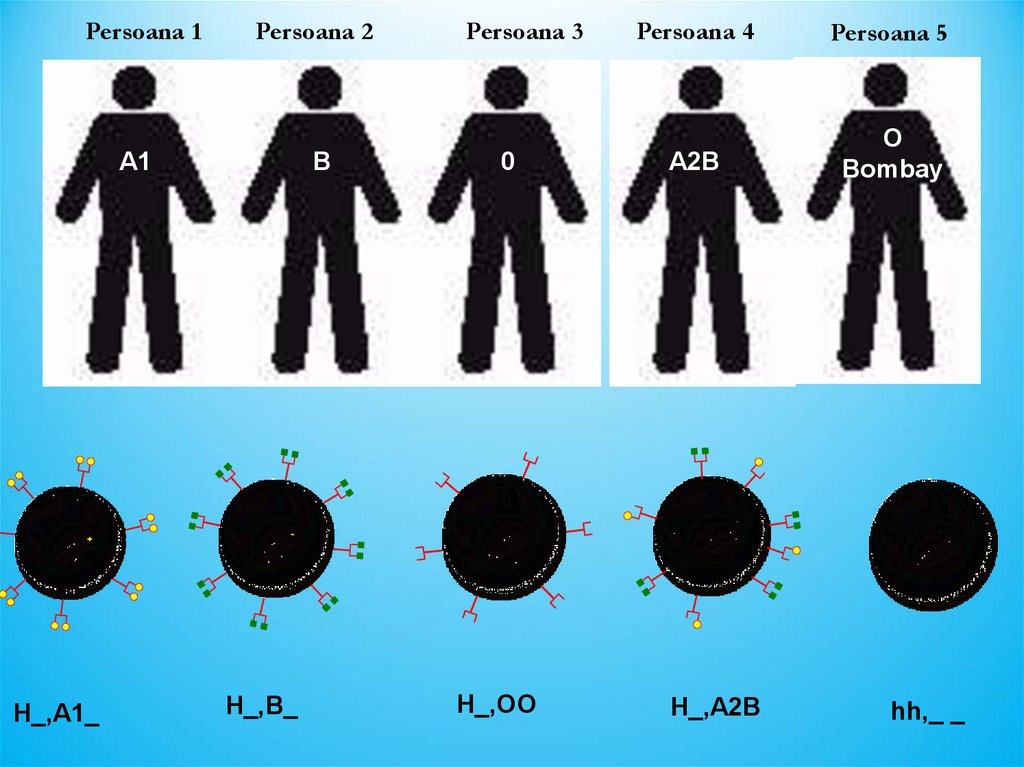
Persoana 1Persoana 2
Persoana 3
Persoana 4
A1, Rh+
B, Rh-
0, Rh+
A2B, Rh+
Fenotip:
Ag A1
Ag D
Ag B
Ag B
Ag H
Ag D
Ag A2
Ag D
Genotip:
A1_ D_
B_ dd
OO D_
A2B D_
30.
O Bombayprecursor
hh
Gena B
Gena A1
Gena A2
Gena O
Gena Se
O
Нет ни антигенов АВО, ни
антигенов Н на
поверхности эритроцитов
31.
Persoana 1Persoana 2
A1
+
H_,A1_
B
Persoana 3
0
Persoana 4
Persoana 5
A2B
O
Bombay
-
H_,B_
H_,OO
H_,A2B
hh,_ _
32.
RhMN
Xg
H
Se
ABO
Rh
Alele
D>d
Genotip
DD
Dd
dd
Xg
MN
ABO
M=N
[(A1>A2)=B] > O Se>se
Xg(a+) > Xg(a-)
MM
MN
NN
OO
A1A1
A1O
A1A2
A2A2
A2O
BB
BO
A1B
A2B
Xg+ Xg+
Xg+ XgXg- Xg-
Se
SeSe
Sese
sese
Xg
Xg+ Y
Xg- Y
H>h
HH
Hh
hh
32
33. [(A1>A2)=B] > O și D>d și M=N și Xg+>Xg-
[(A1>A2)=B] > O și D>d și M=N și Xg+>XgGenotip Fenotip• O(I)
• OO
Genotip
Fenotip
• A1O • A1(II)
• Xg+Xg• Xg+, ♀
• A1A2 • A1(II)
• Xg-Xg• Xg-, ♀
• A2O • A2(II)
• Xg+Y
• Xg+, ♂
• B(III)
• BO
• MN
MN
A1B(IV)
• A1B
• MM
M
A2B(IV)
• A2B
• NN
• N
Rh+
• DD
• A1BDdMM Xg-Y • A1B,Rh+,M, Xg-,♂
• Rh+
• Dd
• BOddMN Xg+Xg-• B,Rh-,MN, Xg+, ♀
• Rh• dd
• OODDNN Xg+Y • O,Rh+,N, Xg+,♂
34.
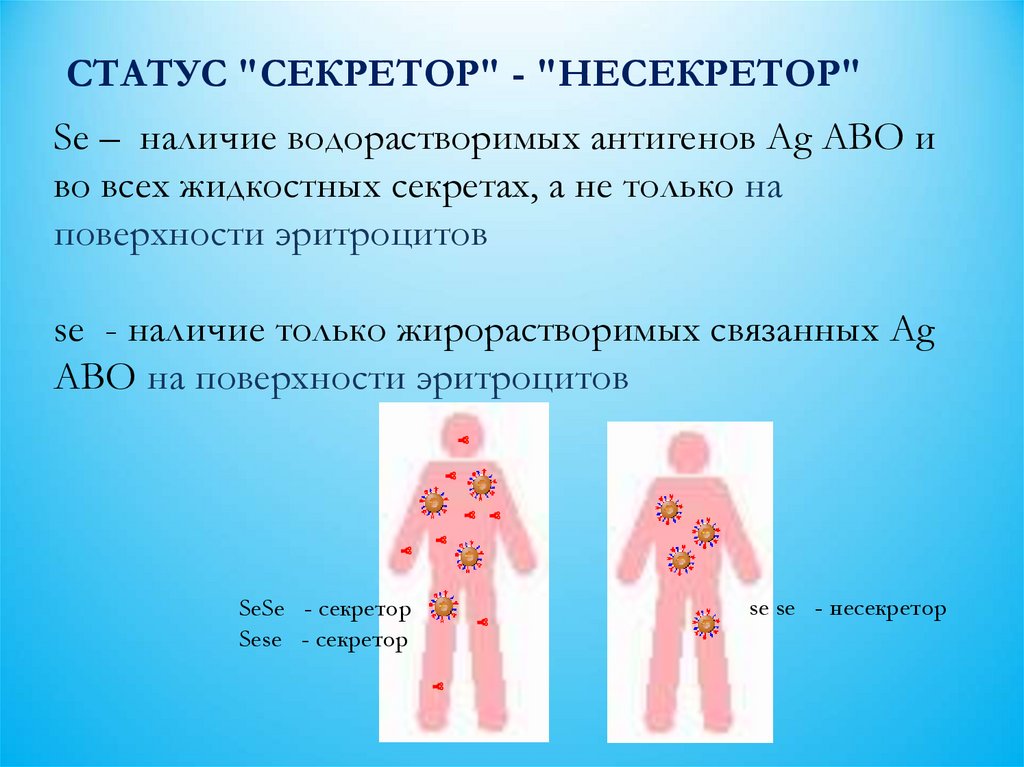
СТАТУС "СЕКРЕТОР" - "НЕСЕКРЕТОР"Se – наличие водорастворимых антигенов Ag АВО и
во всех жидкостных секретах, а не только на
поверхности эритроцитов
se - наличие только жирорастворимых связанных Ag
АВО на поверхности эритроцитов
SeSe - секретор
Sese - секретор
se se - несекретор
35.
Группы крови в системе MN• В этой системе есть несколько фенотипов:
M, N, и MN, которые определяются
кодоминантными аллелями M и N
• Возможные генотипы : MM, NN, MN.
• Знание этой системы важно, в основном, для
судебной медицины.
36.
Характерные отличия иммунной системынесекреторов
- чаще возникают различные воспалительные процессы;
- больше степень риска возникновения сахарного диабета
обоих типов;
- меньшая способность противостоять бактериям;
- чаще встречаются различные аутоиммунные
заболевания;
- повышенный риск рецидивирующих инфекций
мочевыводящих путей;
- чаще наблюдаются заболевания сердечно-сосудистой
системы.
37. Какой фенотип имеет человек с таким генотипом: Hh, B0, dd, MM, Sese
• A1B, Rh-, MN, secretor• B, Rh-, M, secretor,
• A1B, Rh-, M, non-secretor,
• B, Rh+, MN, non-secretor,
• B, Rh+, M, secretor,
38. Какой генотип будет у человека с фенотипом Rh+, A1B, MN, Secretor?
• A1B, dd, MN, sese,• BB, dd, MM, SeSe,
• A1B, Dd, MN, Sese,
• A1B, Dd, MN, Sese,
• A1B, Dd, MM, Sese,
39.
Какой Ag имеет человек наэритроцитах со следующим генотипом:
Hh A1B Dd Sese NN Xg(a+)Y A1, B, N, D, Se, Xg,
Какой Ag имеет человек на
эритроцитах со следующим генотипом:
A1, B, N, D, Se, Xg,
hh A1B Dd Sese NN Xg(a+)Y
Какой Ag имеет человек на
эритроцитах со следующим генотипом:
Hh A2O dd Sese MM Xg(a+)Xg(a-)
A2, M, D, Se, Xg,
40.
Какой Ag имеет человек на эритроцитах соследующим генотипом: Hh OO Dd Sese
H, O, D, Se,
41.
Какой генотип будет учеловека с группой крови А1
Какой генотип будет у
человека с группой крови А1В
A1A1
A10
A1A2
A1B
A2B
A1A1
A10
A1A2
A1B
A2B
42.
Типы взаимодействия геновАллельные
Неаллельные
• Полное доминирование
• Эпистаз
• Неполное доминирование
• Кодоминирование
• Сверхдоминирование
• Аллельная
комплементарность
• Генная
комплементарность
• Эффект положения
• Полимерия
Аллельное
исключение
• Гемизиготность
• Геномный импринтинг
43.
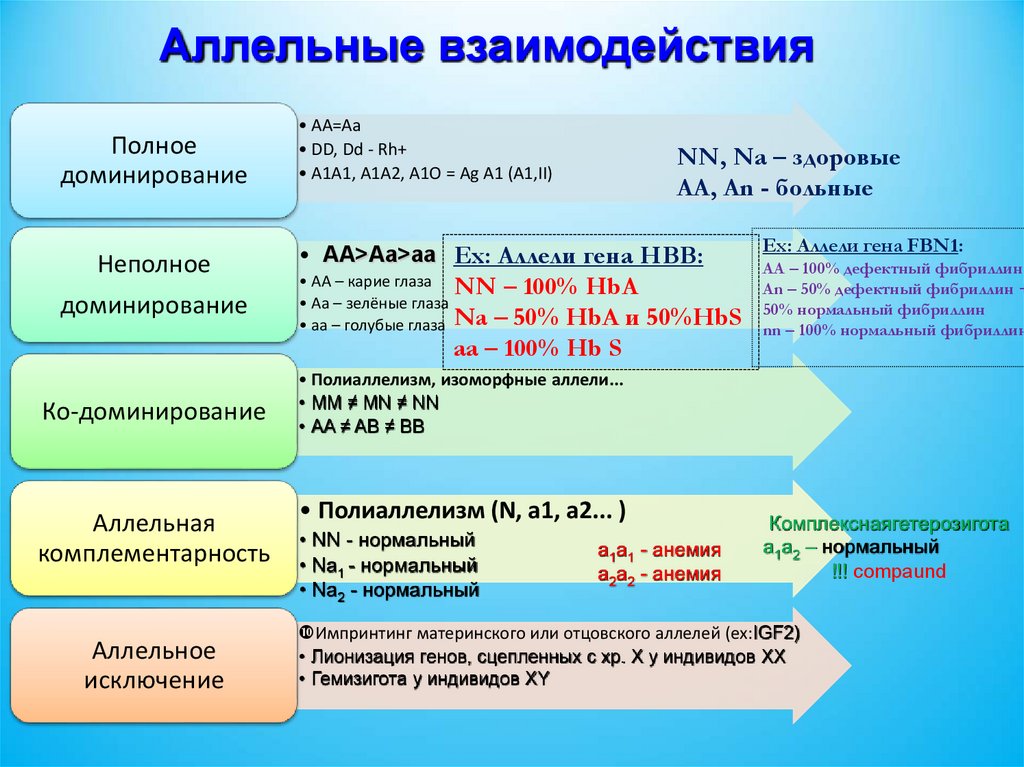
Аллельные взаимодействияПолное
доминирование
Неполное
доминирование
• AA=Aa
• DD, Dd - Rh+
• A1A1, A1A2, A1O = Ag A1 (A1,II)
NN, Na – здоровые
AA, An - больные
• AA>Aa>aa Ex: Аллели гена HBB:
• AA – карие глаза NN – 100% HbA
• Aa – зелёные глаза
• aa – голубые глаза Na – 50% HbA и 50%HbS
aa – 100% Hb S
Ко-доминирование
Аллельная
комплементарность
Аллельное
исключение
Ex: Аллели гена FBN1:
AA – 100% дефектный фибриллин
An – 50% дефектный фибриллин +
50% нормальный фибриллин
nn – 100% нормальный фибриллин
• Полиаллелизм, изоморфные аллели...
• MM ≠ MN ≠ NN
• AA ≠ AB ≠ BB
• Полиаллелизм (N, a1, a2... )
• NN - нормальный
• Na1 - нормальный
• Na2 - нормальный
a1a1 - анемия
a2a2 - анемия
Комплекснаягетерозигота
a1a2 – нормальный
!!! compaund
Импринтинг материнского или отцовского аллелей (ex:IGF2)
• Лионизация генов, сцепленных с хр. X у индивидов XX
• Гемизигота у индивидов XY
44.
Неаллельные взаимодействия• Разные гены с разными локусами кодируют разные белки
• Белковые продукты взаимодействуют в формированииHBA + HBB =
Генная
FBN1+FBN2=
сложного признака
комплементарность
COL1A1+COL1A2=
• Мутации – приводят к сходным фенотипам
PKD1+PKD2=
Эпистаз
Эффект позиции
Аддитивная
полимерия
• Разные гены с разными локусами кодируют разные белки
• Белковые продукты активируют друг друга в цепочке, F8 F9
PAH Tyr
образуя сложный признак
PAX3 др. гены
• Мутации - приводят к сходным фенотипам
• Разные гены на соседних локусах = гаплотип, с общими
регуляторными элементами, кодируют разные белки crs11: IGF2 H19
• Мутации гена - могут вызывать фенотипические
crs1: C D E
изменения в соседнем гене
• t, inv crs – может модифицировать экспрессию
задействованных генов
• Разные гены, разные локусы кодируют один и тот же
белок/белки с похожими функциями.
• Продукты нескольких генов суммируются для
определения количественного признака.
• Количество меланина и цвет кожи
• АД, IQ, рост, масса тела
A-код.меланин
B-код.меланин
C-код.меланин
AABBCC
AaBbCc
aaBBCc
Aabbcc
45. Взаимодействия аллельных генов
• Аллельные гены определяют моногенные признаки ихарактеризуются биаллельной экспрессией.
• Сочетания разных вариантов аллелей обеспечивает
генотипическое и фенотипическое разнообразие
индивидумов.
• У гомозигот аллельные гены одинаковы, а у
гетерозигот – разные.
46. Взаимодействия аллельных генов
• Аллельные гены могут быть доминантными ирецессивными по своему проявлению в фенотипе, в то
время как на молекулярном уровне различить их
трудно, так как оба гена кодируют один белок.
- Доминанатные гены характеризуются активным
генопродуктом и способностью транскрибироваться в
определенное количество мРНК.
- Рецессивные гены транскрибируются меньше или их
генопродукт неактивен.
47.
Аллельные взаимодействияПолное
доминирование
Aa = AA
Rh
Секретор
Xg
Неполное
доминирование
AA>Aa>aa
HbA HbA – здоровый
HbA HbS – анемия
HbS HbS –летальный
исход
Аллельное
исключение
Ig
Х-сцепленные гены
Кодоминирование
MM ≠ MN ≠ NN
MN
AB
HLA
Гаптоглобины
Аллельная
комплементарность
AA – норма AA1 -норма
AA2 – норма A1A2 - норма
A2A2 – мутант A1A1 – мутант
48.
Полное доминированиеНаследование фактора Rh
D – Rh+
d – Rh-
D – доминантный аллель
d – рецессивный аллель
Р
DD x dd
P
Dd x dd
P
Dd x Dd
G
D
d
G
Dd
d
G
Dd Dd
Dd – 100%
F1
Dd – 50%
dd – 50%
F1
DD – 25%
Dd – 50%
dd – 25%
F1
49. Неполное доминирование
Фенотипическиaa <Aa < AA
Exemple:
AA – кучерявые волосы
Aa – волнистые волосы
aa – прямые волосы
• AA – здоров
• AS – легкая форма анемии
• SS – летальная форма анемии
• NN – здоров
• Na – образование песка в почках
• aa – образование цистеиновых камней
50. Кодоминирование
• Гены А и В в системе АВ0• Гены М и N в системе MN
• Гены Hp1 и Hp2
51. Аллельная комплементарность
• A > A1 ; A > A2 ; A1= A2А – нормальный доминантный аллель
– A1 неоморфный рецесс. патол. аллель, кодир.один
белковый домен
– A2 неоморфный рецесс. патол. (но с другой мутацией)
аллель, кодир.другой белковый домен
• Примеры:
AA – норма норм Hb
AA1 – норма норм Hb
AA2 - норма норм Hb
A1A1 - мутант дефектный Hb
A2A2 - мутант дефектный Hb
A1A2 - норма норм Hb – у человека два патологических рецесс.
аллеля, но с разными мутациями, а вместе они нивелируют патологическое
проявление и получается нормальный фенотип по данному признаку. Такая
гомозигота, которая состоит из, вроде 2 патологич. Аллелей, т.е. должна
была бы быть гомозиготой, но состоит из 2 рецесс. патол. аллелей с
разными мутациями, называется сложной гетерозиготой - compound
52. Аллельное исключение
• Гемизиготность– XHY
– XhY
Инактивация
Инакт. Xh
H
h
X X
норм. коагуляция
Инакт.X
H
h
X X
гемофилия
H
Обеспечивает разнообразие
синтезируемых в организме человека
антител
53.
Неаллельные взаимодействияАдитивная
полимерия
Эпистаз
Генная
комплементар
ность
Цвет кожи:
Бомбейский
фенотип
Образование
гемоглобина
A1 –меланин
A2 – меланин
A3 –меланин
a –рецессивн.
аллель
A1A1A2aA3A3
A1aA2aA3a
IQ
Вес тела
Несекретор
Слух
Эффект
положения
CDE
(фактор
Rh )
54.
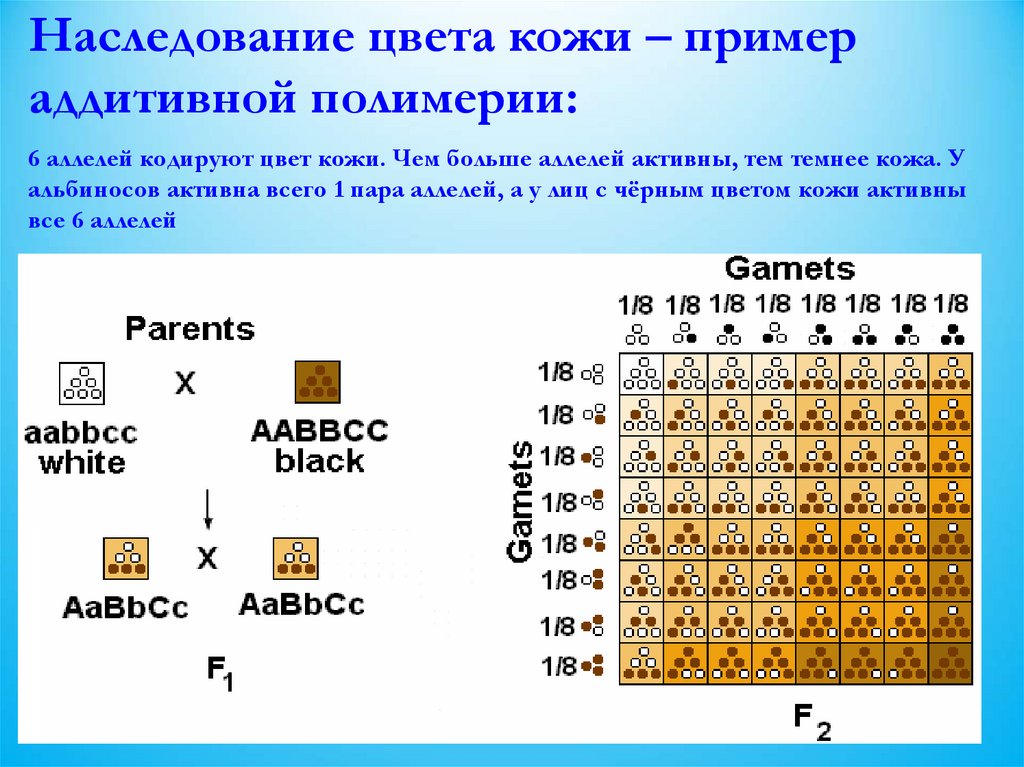
Наследование цвета кожи – примераддитивной полимерии:
6 аллелей кодируют цвет кожи. Чем больше аллелей активны, тем темнее кожа. У
альбиносов активна всего 1 пара аллелей, а у лиц с чёрным цветом кожи активны
все 6 аллелей
55.
Гауссово распределение в популяции56.
Эпистаз – явление, при котором доминантный(рецессивный) ген одной аллельной пары подавляет
действие другого неаллельного гена.
Генотип 00 Hh – гр. 0 (присутствует антиген H)
Генотип B0 hh – фенотип Bombay (отсутствие
антигенов системы AB0)
57.
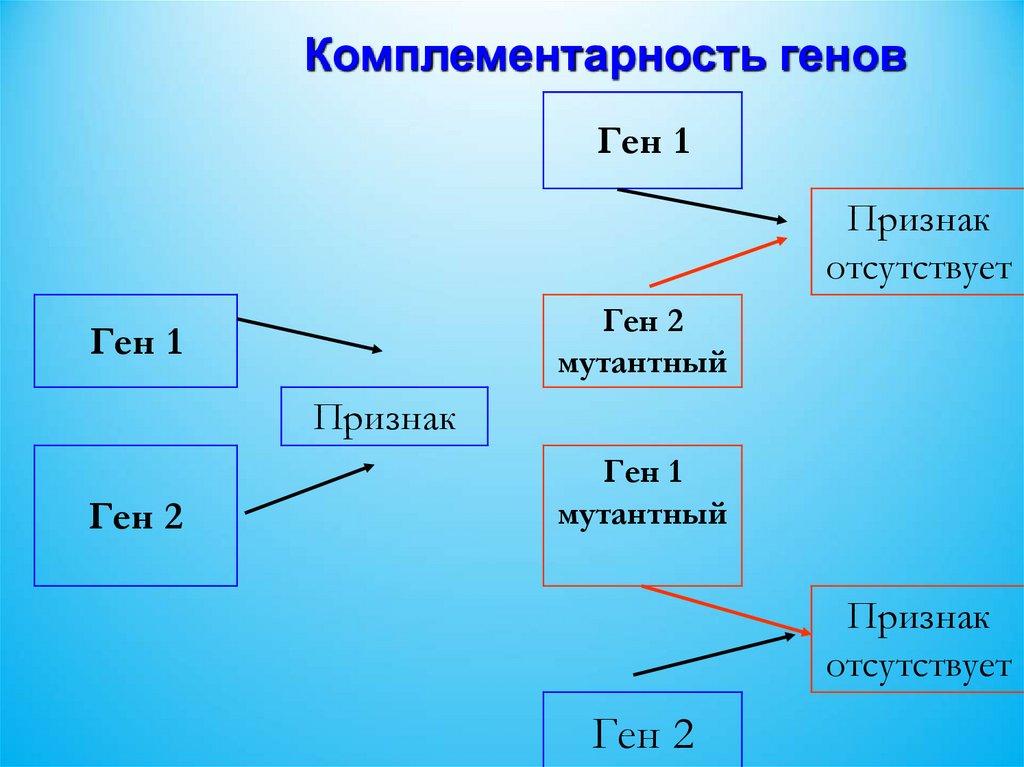
Комплементарность геновГен 1
Признак
отсутствует
Ген 2
мутантный
Ген 1
Признак
Ген 2
Ген 1
мутантный
Признак
отсутствует
Ген 2
58.
Примеры комплементарного действия геновНормальный гемоглобин
Ген α-глобина – в хр.16
+
Ген В-глобина –в хр.11
= нормальный гемоглобин
59.
Примеры комплементарногонеаллельного взаимодействия генов
Наследование слуха у человека контролируется
двумя неаллельными комплементарными
генами: D и E
DDEE, DDEe, DdEe, DdEE –
нормальный слух
ddEE, ddEe, DDee, Ddee, ddee –
слух отсутствует
60.
Эффект положения - нарушение или изменениеуровня экспрессии гена в результате изменения его
местоположения
в
структуре
генома,
но
не
сопровождающееся структурной патологией или мутацией.
При этом ген сохраняется как транскрипционная единица
с
соответствующей
промоторной
нетранслируемым участком.
областью
и
3’-
61.
Патологические моногенные признаки1. AD
с. Марфана АА, An и nn
2. AR
Фенилкетонурия aa и NN, Na
1. XD
c. Alport XAXA ,XAXn, XAY, XnXn ,XnY
1. XR
Гемофилия А XaXa , XaY, XNXN, XNXa ,XNY
2. Митохондриальные
MELAS
62.
Моногенные патологииAA, An, nn
AD
* Marfan (FBN1)
* Boala polichistica renala (PKD1)
Hipercolesterolemia familiala (LDLR)
Hutington (HD sau Htt)
* Waardenburg (PAX3)
* Li-Fraumeni (TP53)
* Osteogeneza imperfecta (COL1A1)
* Beckwith-Wiedemann (IGF2, ♂)
XD
* Alport (COL4A5)
* X-FRA (FMR1)
XAXA, XAXn XAY
XnXn XnY
aa, NN,Na
AR
Fenilcetonuria (PAH)
* Mucoviscidoza (CFTR)
Alfa-talasemia (HBA)
Beta-talasemia, Anemia S (HBB)
XR
* DMD (DMD)
Hemofilia A (F8)
Hemofilia B (F9)
первичная плейотропия* , а остальные – вторичная плейотропия
XaXa
XaY
XNXN
XNXa
XNY
63.
Генетические феномены, которымиобусловлено разнообразие проявления
патологических фенотипов при генетических
моногенных патологиях
64. Генетические феномены (их нужно учитывать при анализе задач)
1. Импринтинг Экспрессия, зависящая от родительского происхождения гена2. Антиципация патологии
Нестабильность некоторых генов, вследствие динамических мутаций
3. Пенетрантность
Полная
Неполная
4. Генетическая гетерогенность: разные мутации, но сходный фенотип
Аллельная
Неаллельная ( синоним – локусная)
5. Вариабельная экспрессивность патологии Различные формы заболевания (по степени тяжести и т.д.)
65.
1. Клиническийполиморфизм
2. Антиципация
3. Плейотропия
Полная и неполная
пенетрантностьь зависит от:
возраста
пола
Родительского
происхождения
Факторов среды
Пенетрантность
Frecvența unei
gene dominante de
a se manifesta la An
% An – bolnavi vs
% An sănătoși
Вариабельная
экспрессивность
Разная степень
проявления/
полные и неполные
проявления
синдрома
Завистит от:
• генотипа
• пола
• среды
• возраста
• ткани
Взаимодейст
вия между
генами
-Аллельные
Неаллельные
Heterogenitatea
alelică / nealeică
diferite mutații –
manifestări
asemănătoare
66.
s.Marfan(An)
Поликистоз
почек (An AA)
Различные
мутации в гене
FBN1 и дефект
фибриллина 1
Различные
мутации в гене
PKD1 или
мутации в PKD2
или в PKD3 ...
>36 CAG в гене
Htt = дефектный
белок гентингтин
Различные
мутации в гене
COL1A1 или
COL1A2 = дефект
коллагена 1
скелетные,
глазные и
сердечнососудистые
аномалии
почечные и внепочечные
кисты!!, почечная
недостаточность,
артериальная
гипертензия, пролапс
митрального клапана,
аневризмы
апоптоз нейронов и
прогрессирующая
дегенерация
головного мозга
при психических
расстройствах и
деменции
аномалии
скелета, хрящей,
костей, кожи,
склеры
Полные и неполные
формы синдрома,
но больны все
гетерозиготы
тяжелые и
менее тяжелые
формы с
началом после
30 лет
Различные формы с
началом после 50
лет или ранее, в
зависимости от
числа повторов CAG
Аллельная гетерогенность
Аллельная гетерогенность для
гена PKD1 в частности и
неаллельная (PKD1, PKD2, PKD3 )
Аллельная гетерогенность
Вторичная плейотропия
Первичная плейотропия
Huntington (An)
Osteogenesis
imperfecta (An)
Вариабельная
экспрессивность
Первичная плейотропия
Expresivitate variabilă
Вариабельная экспрессивность
Полная пенетрантность
Пенетрантность, зависимая от
Пенетрантность, зависимая от
возраста, Антиципация
Тяжелые и менее
тяжелые формы в
зависимости от
мутаций и факторов
окружающей среды
Аллельная гетерогенность для гена
COL1A1 в частности и
неаллельная(COL1A1, COL1A2 )
Первичная плейотропия
Expresivitate variabilă
Полная пенетрантность
67.
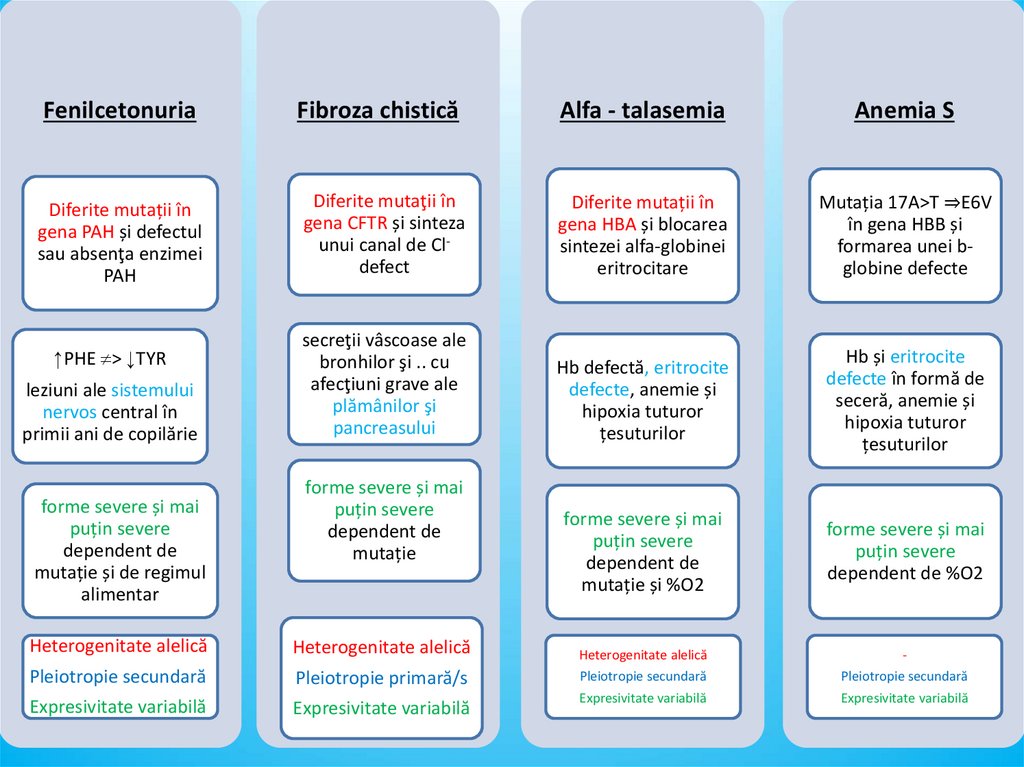
FenilcetonuriaFibroza chistică
Alfa - talasemia
Anemia S
Diferite mutații în
gena PAH și defectul
sau absenţa enzimei
PAH
Diferite mutaţii în
gena CFTR și sinteza
unui canal de Cldefect
Diferite mutații în
gena HBA și blocarea
sintezei alfa-globinei
eritrocitare
Mutația 17A>T ⇒E6V
în gena HBB și
formarea unei bglobine defecte
secreţii vâscoase ale
bronhilor şi .. cu
afecţiuni grave ale
plămânilor şi
pancreasului
Hb defectă, eritrocite
defecte, anemie și
hipoxia tuturor
țesuturilor
Hb și eritrocite
defecte în formă de
seceră, anemie și
hipoxia tuturor
țesuturilor
forme severe și mai
puțin severe
dependent de
mutație și %O2
forme severe și mai
puțin severe
dependent de %O2
↑PHE > ↓TYR
leziuni ale sistemului
nervos central în
primii ani de copilărie
forme severe și mai
puțin severe
dependent de
mutație și de regimul
alimentar
forme severe și mai
puțin severe
dependent de
mutație
Heterogenitate alelică
Heterogenitate alelică
Heterogenitate alelică
-
Pleiotropie secundară
Pleiotropie primară/s
Pleiotropie secundară
Pleiotropie secundară
Expresivitate variabilă
Expresivitate variabilă
Expresivitate variabilă
Expresivitate variabilă
68. Плейотропия – явление, при котором один ген определяет формирование нескольких признаков
ПлейотропияПервичная
Вторичная
Признак 1
ген
Признак 2
Признак 3
Пример: синдром Марфана
Признак 1
ген
Признак 3
Признак 2
Пример: муковисцидоз, талласемия
69.
Мутантные аллели различных генов,контролирующих синтез коллагена и
фибриллина, приводят к нарушению
свойств волокнистой соединительной
ткани. Т.к. соединительная ткань - основа
всех органов и тканей, то понятны
множественные влияния этих мутаций на
клиническую картину (фенотип) при таких
наследственных болезнях соединительной
ткани, как, например, синдромы ЭлерсаДанло, Марфана.
70.
Прифенилкетонурии
нарушается
обмен
фенилаланина, в результате чего не синтезируется
тирозин,
уменьшается
или
прекращается
образование меланина, что ведёт к гипопигментации
кожи, волос и радужки. Накопление патологических
метаболитов ведет к нарушению процессов развития
и
работы
нервной
системы
(повышенная
возбудимость,
тремор,
судорожные
припадки,
умственная отсталость). В основе всех этих
множественных проявлений
лежит первичный
эффект недостаточности (или отсутствия) активности
фенилаланингидроксилазы.
71. Генетическая гетерогенность
• явление, когда мутации в разных локусах илиразные мутации в одном локусе имеют сходное
фенотипическое проявление.
• Различают два типа генетической гетерогенности:
1. Локусная гетерогенность
2. Аллельная гетерогенность
72.
1. Локусная гетерогенность:- сходный фенотип обусловлен действием
разных неаллельных генов;
- обозначается термином генокопия;
73. Примеры болезней человека с локусной гетерогенностью
БолезньХарактеристика
Локализация генов
Болезнь Альцгеймера
Прогрессирующее
слабоумие
1,4,19,21
Болезнь Шарко-Мари
Периферическая
нейропатия
1,5,8,11,17,Х
Рак молочной железы
Предрасположенность
к раку в раннем
возрасте
13,17
Синдром ЭлерсаДанлоса
Поражения
соединительной ткани
Х, аутосомы
Пигментный ретинит
Прогрессирующая
ретинопатия и потеря
зрения
3,6,8,19,Х
Несовершенный
остеогенез
Поражения костей
7,17
74.
2. Аллельная гетерогенность:- мутации разных аллелей одного локуса проявляются
сходным фенотипом, при этом варьирует интенсивность
проявления,
- основой является множественный аллелизм,
Примеры: мукополисахаридозы, мышечная дистрофия
Дюшена, болезнь Беккера, гликогенозы и др.
75. Mutaţii diferite – fenotipuri identice = heterogenitate genetică (alelică și non-alelică)
• Heterogenitate alelică– Mutaţii diferite ale aceluiaşi
locus
– Variante alelice multiple
– Fenotipuri asemănătoare
• Exemple
– Mutaţia amorfă lipsa FVIII
Hemofilia A
– Mutaţia neomorfă F VIII
defect hemofilia A
Heterogenitate non-alelică (de
locus)
Mutaţii diferite ale diferitor
gene nealele
Datotită acţiunei
complimentare sau epistatice
a genelor
Determină fenotipuri
asemănătoare
Exemple
Mutaţia genei α- globinei (crs
16) Hb defectă anemie
Mutaţia genei β- globinei (crs
11) Hb defectă anemie
76.
Мутации в гене PKD1 или PKD2 могут вызыватьаутосомно-доминантную поликистозную болезнь почек;
Эти гены обеспечивают инструкции для создания белков
полицистина 1 и полицистина 2, участвующих в
передаче химических сигналов. Эти два белка работают
вместе,
способствуя
нормальному
развитию,
организации и функционированию почек и другого
эпителия. Мутации в гене PKD1 или PKD2 приводят к
образованию
тысяч
кист,
которые
нарушают
нормальные функции почек и других органов. Люди с
мутациями в гене PKD2, особенно женщины, обычно
имеют менее тяжелую форму заболевания, чем люди с
мутациями PKD1. Признаки и симптомы, включая
снижение функции почек, как правило, появляются
позже во взрослом возрасте у людей с мутацией PKD2.
77.
Более 500 мутаций в гене PAH были выявлены у людей сфенилкетонурией (ФКУ/PKU). Большинство этих мутаций
приводят
к
синтезу
дефектной
фенилаланингидроксилазе.
Например,
самая
распространенная мутация F508. Признаки и симптомы
фенилкетонурии варьируются от легких до тяжелых.
Наиболее тяжелая форма этого состояния известна как
классическая
фенилкетонурия.
У
детей
с
фенилкетонурией обычно нарушены оба аллеля, они
выглядят нормальными в течение первых месяцев после
рождения, и без лечения у них развивается постоянная
умственная отсталость. Часто встречаются судороги,
задержка развития, поведенческие проблемы и
психические расстройства.
78. Вариабельная клиническая экспрессия (проявление) при мутациях в одном и том же гене
• Различная степень или форма проявленияодного и того же гена у разных людей:
• Зависит от генотипа (например, Rh+)
• В зависимости от факторов окружающей
среды (например, АДГ-аза)
• Половая зависимость (например, ген
аллопеции)
• Тканевая специфичность (например, ген
ангиоматоза)
• Зависит от стабильности генов (например,
динамические мутации и антиципация)
79. Penetranţa genei
• Capacitatea genei dominante de a se manifesta lahetozigoţi (An):
a) 100% An - caracter dominant – penetranţă completă
b) 60% An - caracter dominant şi
40 % An caracter recesiv – penetranţă incompletă
c) 100% An - ♀ - caracter dominant şi 0% An - ♂ - caracter recesiv –
penetranţă incompletă, limitată de sex
• Fenomen genetic determinat de:
–interacţiuni genice
–factori de mediu
–Sex
–Vîrstă
–Origine parentală
–…..
HhA1B = A1B
hhA1B = O, Bombay
80. Моноаллельная экспрессия
• Проявление только одного аллеля из парыаллельных генов
• Гены с моноаллельной экспрессией у человека:
- гены, локализованные только в хромосомах Х
или Y у мужчин,
- гены хромосом Х у женщин,
- гены признаков, контролируемых или
ограниченных полом (ген плешивости, лактации и
др),
- импринтированные гены.
81.
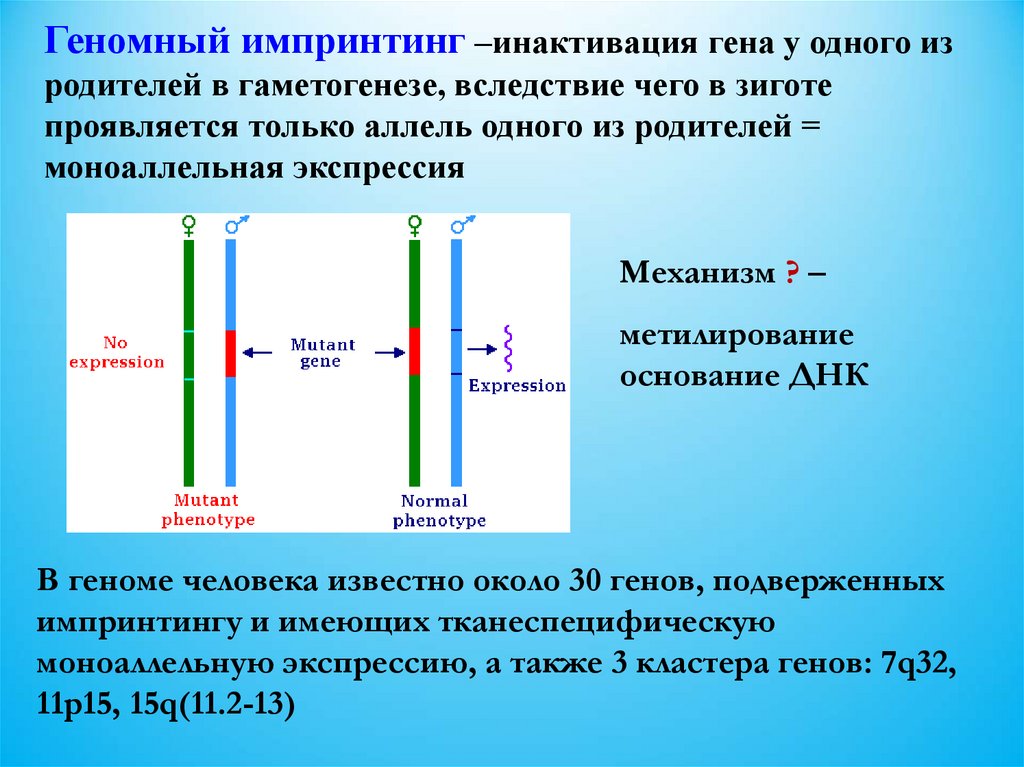
Геномный импринтинг –инактивация гена у одного изродителей в гаметогенезе, вследствие чего в зиготе
проявляется только аллель одного из родителей =
моноаллельная экспрессия
Механизм ? –
метилирование
основание ДНК
В геноме человека известно около 30 генов, подверженных
импринтингу и имеющих тканеспецифическую
моноаллельную экспрессию, а также 3 кластера генов: 7q32,
11p15, 15q(11.2-13)
82. Примеры геномного импринтинга у человека
СиндромЛокализация
гена
Импринт.
аллель
Активный Клинические признаки
аллель
С.ПрадераВилли
15q11q13
М
О
Гипотония, ожирение,
умственное отставание
С.Ангельмана
15q11q13
О
М
Нарушения речи,
микроцефалия,
гипопигментация кожи
С.Беквита
11p11.5
М
О
Гигантизм, нарушения
внутренних органов
С.СилвераРассела
7p12p14
О
М
Отставание в развитии,
черепно-лицевые
аномалии
С.Гентингтона
4p16.3
М
О
Прогрессирующая
дегенерация базальных
ганглионов,
двигательные
нарушения
83.
Болезнь Гентингтона – это прогрессирующеенейродегенеративное заболевание с аутосомнодоминантной передачей. Более 36 повторений
тринуклеотидного микросателлита в гене HD
вызывают синтез дефектного белка гентингтина,
вызванного последовательностью polyGLN,
которая накапливается в нейронах, вызывая
апоптоз нейронов и прогрессирующую
дегенерацию мозга. Проявление заболевания
(начало и тяжесть) зависит от количества
тринуклеотидных повторов.
Mutații???
Fenomene?
Expresivitatea
Heterogenitatea
Anticipația
Pleiotropia
Penetranța
84.
Ген HD или HTT (4p16.3) кодирует белок гентингтин. Молекулярныйуровень – синт. дефектный белок hd. Клеточный уровень - апоптоз
нейронов. Организменный – дегенерация головного мозга
Охарактеризуйте мутации гена HD?-генная-доминантная- неоморфная
(т.к. белок ДЕФЕКТНЫЙ) - экзонная-динамическая Плейотропия Вторичная: ген HD затрагивает дегенерация головного мозга, а
остальные клинические проявления – - это множественные
осложнения патологии головного мозга Гетерогенность? – Аллельная
гетерогенность, т.к. разные мутации, разное количество повторов в
одном гене - гене HD, приводят к болезни Гентингтона.
Экспрессивность? – вариабельная - У разных пациентов болезнь H
проявляется по-разному, разная степень тяжести. Прогноз? Т.к. это
Динамическая мутация, то у детей больных родителей болезнь Н будет
проявляться раньше и тяжелее = антиципация. Пенетрантность?
Полная, зависимая от возраста: у всех Аn будет проявляться болезнь
Н... Методы выявления носителей мутаций и диагностики больных?
ПЦР-амплификация / Саузерн-блот / дидезокси-секвенирование ДНК
по Сэнгеру.
85.
Исследователи выявили более 1300 мутаци в гене FBN1,которые вызывают синдром Марфана, состояние,
поражающее соединительную ткань. Большинство мутаций,
вызывающих синдром Марфана, превращают одну
аминокислоту в другую в белке фибриллин-1 – это приводит к
появлению аномального белка фибриллина-1, который не
может функционировать должным образом. Синдром
Марфана — это состояние, которое поражает
соединительную ткань во многих частях тела, таких как кости,
связки, мышцы, кровеносные сосуды и сердечные клапаны.
Признаки и симптомы синдрома Марфана сильно
различаются по тяжести, времени начала и скорости
прогрессирования.
Мутации???
Fenomene?
Экспрессивность
Гетерогенность
Антиципация
Плейотропия
Пенетрантность
86. Менделирующие и неменделирующие признаки
МенделирующиеМоногенные
Биаллельный
детерминизм
Доминантные или
рецессивные (AD,
AR, XD, XR)
В популяции –
бимодальное
распределение
Неменделирующие
Моногенные
Моноаллельный
детерминизм
Неполное
доминирование
Кодоминирование
Аллельная
комплементарность
Полигенные
Мультифакториальные
Факторы окружающей
среды влияют на их
экспрессию
Полигенный признак
или полигенная
патология, ни
доминантны, ни
рецессивны, а
передаются они только
в виде
предрасположенности
87.
Нормальные или патологические признакиМенделирующие или неменделирующие?
Мендел. – законы Менделя могут быть применены для
расчета вероятности (риска) наследования патологии,
если AD / AR / XD / XR
Немендел. – законы Менделя не могут быть применены
для расчета вероятности (риска) наследования патологии
88. S.Marfan AD An больны nn здоровы
nnAn
An
nn
nn
An
Мутация
de novo
Неполная
пенетрантность
An
nn
An
nn
An
An
Неполная
пенетрантн
ость
89. Таласемие АР тип наследования aa NN Na
alfa-talasemia beta-talasemaa
aa
Na
Na
a1a1
a2a2
aaBB
AAbb
a1a2 –
25% aa
100% aa
Na
aa
?
aa
NN
?
здоров сложная
гетерозигота –
«compound»
Aa Bb = NaNa
сложная гетерозигота
«compound»? –
фенотип здоров
90.
SanatosBolnav
Na
An
Amprentarea genei N
Interacțiuni nealelice
Vîrsta
Sexul
Factori de mediu
Excepție
alelică
Bolnav
Amprentarea genei A
Interacțiuni nealelice
Vîrsta
Sexul
Penetranță
incompletă
Factori de mediu
Sanatos
91.
DAR…Sanatos
Bolnav
N
a
X X
A
n
X X
Lyonizarea genei N
Interacțiuni genice
Excepție
alelică
etc
Bolnav
Lyonizarea genei A
Interacțiuni genice
etc
Expresie
variabilă
Sanatos
92.
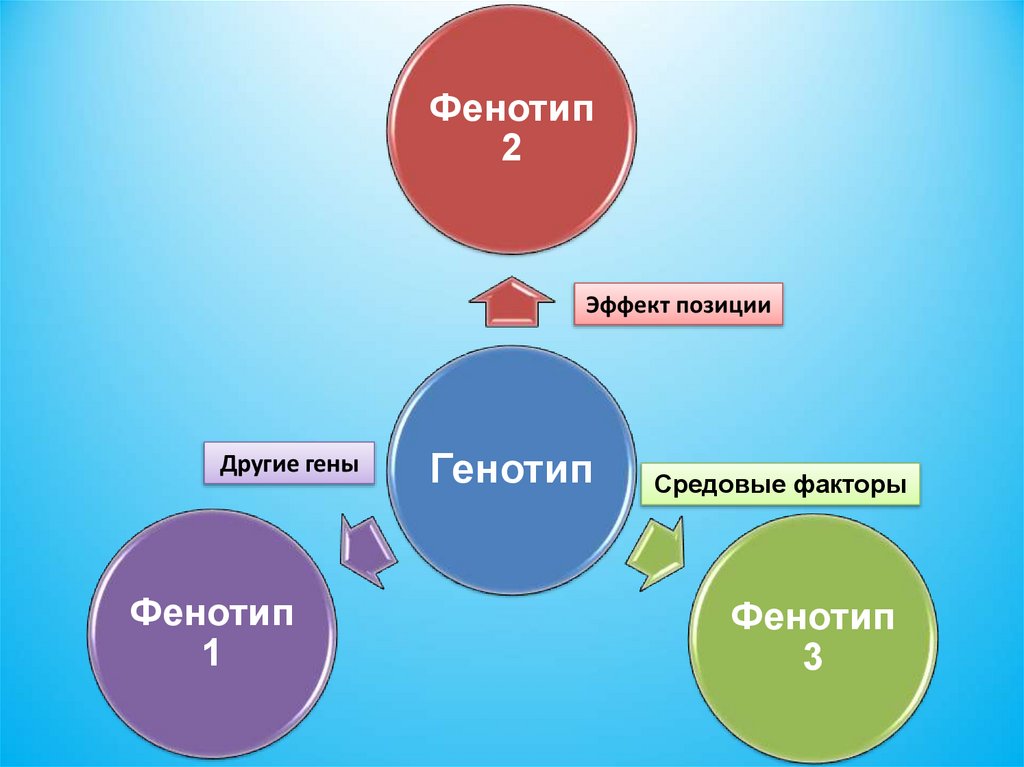
Фенотип2
Эффект позиции
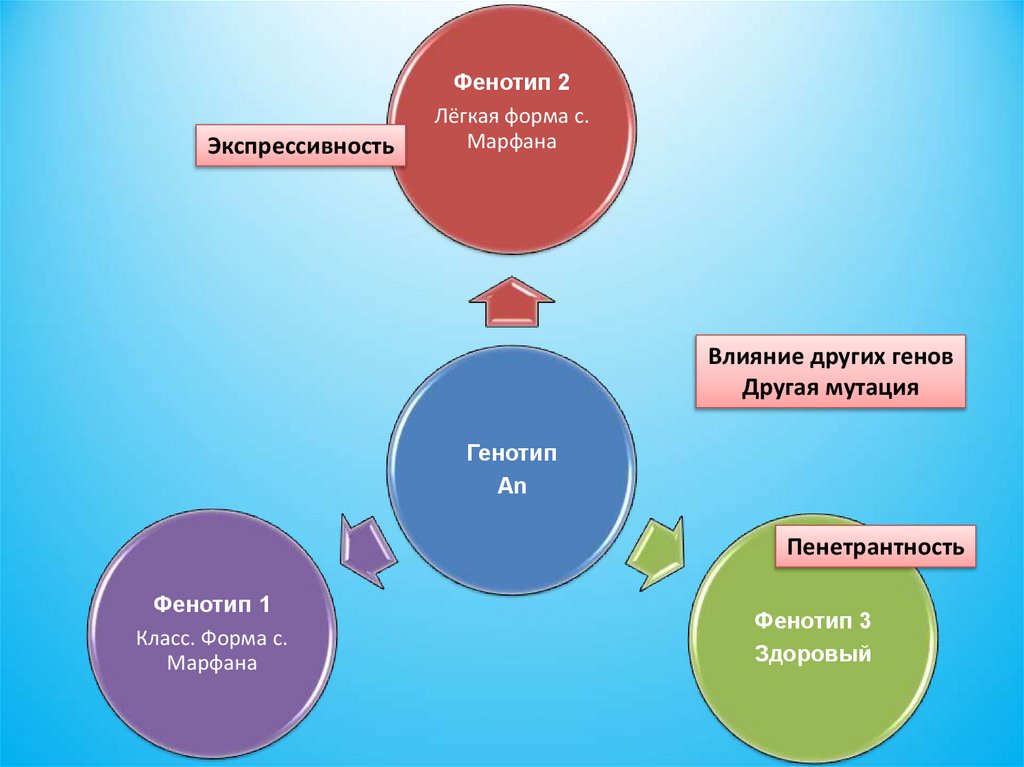
Другие гены
Фенотип
1
Генотип
Средовые факторы
Фенотип
3
93.
ЭкспрессивностьФенотип 2
Лёгкая форма с.
Марфана
Влияние других генов
Другая мутация
Генотип
An
Пенетрантность
Фенотип 1
Класс. Форма с.
Марфана
Фенотип 3
Здоровый
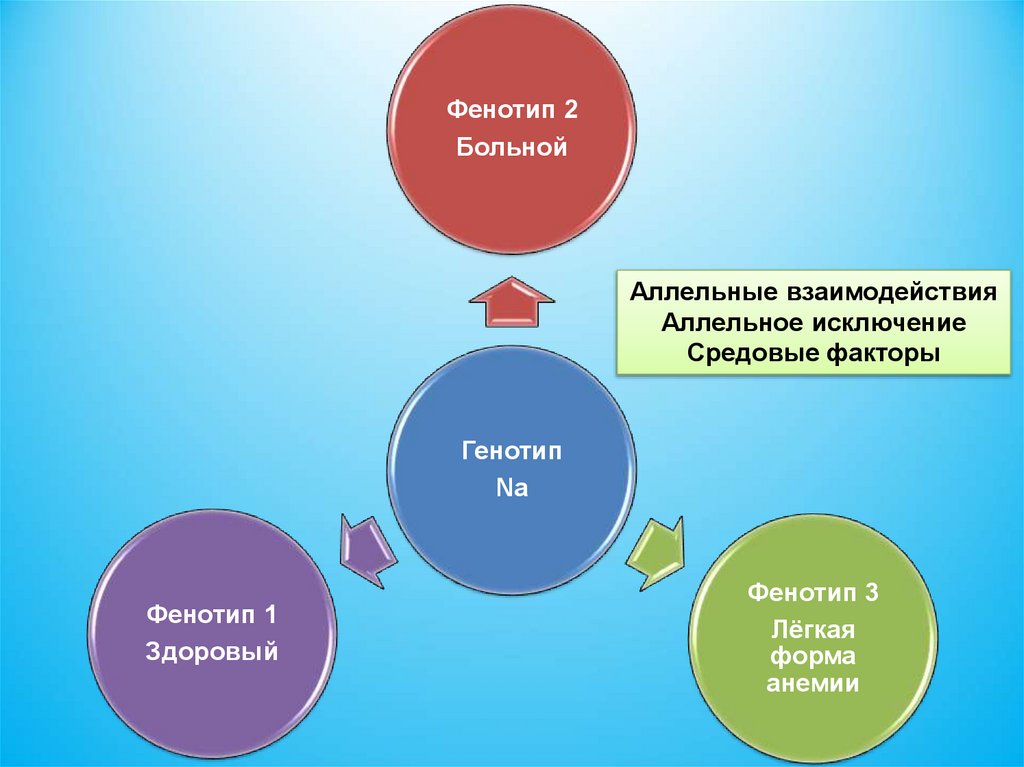
94.
Фенотип 2Больной
Аллельные взаимодействия
Аллельное исключение
Средовые факторы
Генотип
Na
Фенотип 1
Здоровый
Фенотип 3
Лёгкая
форма
анемии
95.
Фенотип2
Комплементарность генов
Эпистаз
Полное доминирование
Эффект позиции
Неполное доминирование
Кодоминирование
Фенотип
1
Генотип
Средовые факторы
Средовые факторы
Фенотип
3
96. Caractere monogenice non-mendeliene
• Mutații în gena IFG2– IGF2 materna
– IGF2 paterna
• Mutații în gena Htt
– 40 expansiuni trinucleotidice
– 70 expansiuni trinucleotidice
– 110 expansiuni trinucleotidice
• Mutații in gena HBA ( aa – bolnavi)
– a1a1
– a2a2
– a1a2
• Mutații în PAX3
– Amorfa, generativă
– Hipermorfa, somatică
97. Caractere monogenice non-mendeliene
• Mutații în gena PKD1 sau PKD2– Polycystin 1 + policystin 2 = receptor membranar funcțional
• Mutații în gena TP53
– An
– An / AA
• Mutații in gena F9
– Mutație in promotor -20
– Mutație in promotor -26
– Mutație în exon 2
• Mutații în mtDNA gene MT-TL1
– Gena maternă
– Gena paternă