












































































 medicine
medicineSimilar presentations:





")




Иммунопатология. Иммунодефициты
1. Иммунопатология
ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ ВЫСШЕГО ОБРАЗОВАНИЯ
«БАШКИРСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ»
МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
Лекция
Иммунопатология
Иммунопатология
Иммунодефициты
Кафедра патофизиологии
Уфа 2017
2.
• Организм человека имеет множество механизмовзащиты от различных вредных агентов: от
неживых веществ, таких, как токсины, химикалии
или инородные частицы, и от живых организмов,
в том числе (и в первую очередь) от возбудителей
инфекционных болезней - вирусов, бактерий,
грибов, простейших и гельминтов. Защиту
осуществляют две системы:
• неспецифическая (сопротивляемость
организма) и
• специфическая (иммунная система)
3.
НЕСПЕЦИФИЧЕСКАЯ ЗАЩИТА ОРГАНИЗМА
• Линии защиты. Сопротивляемость организма складывается из двух
линий защиты. Поверхностные анатомические барьеры (эпителий кожи
и слизистых оболочек), находящиеся на границе внутренней и внешней
среды, - первая линия неспецифической защиты. Физикохимические и биологические свойства эпителия, а также выделяемые
на поверхность эпителия секреторные вещества и клетки не позволяют
патогенам попасть во внутреннюю среду организма.
• Если же патоген преодолевает этот поверхностный барьер и
оказывается во внутренней среде организма, его встречает комплекс
клеточных и гуморальных неспецифических факторов. Это вторая
линия неспецифической защиты, к которой относятся
фагоцитирующие клетки, комплемент, интерфероны, кинины и
некоторые другие вещества, а также естественные антитела
(антигеннезависимые) и естественные киллеры. В совокупности обе
линии защиты составляют врождённые, естественно присущие
организму, т.е. конституциональные факторы.
4.
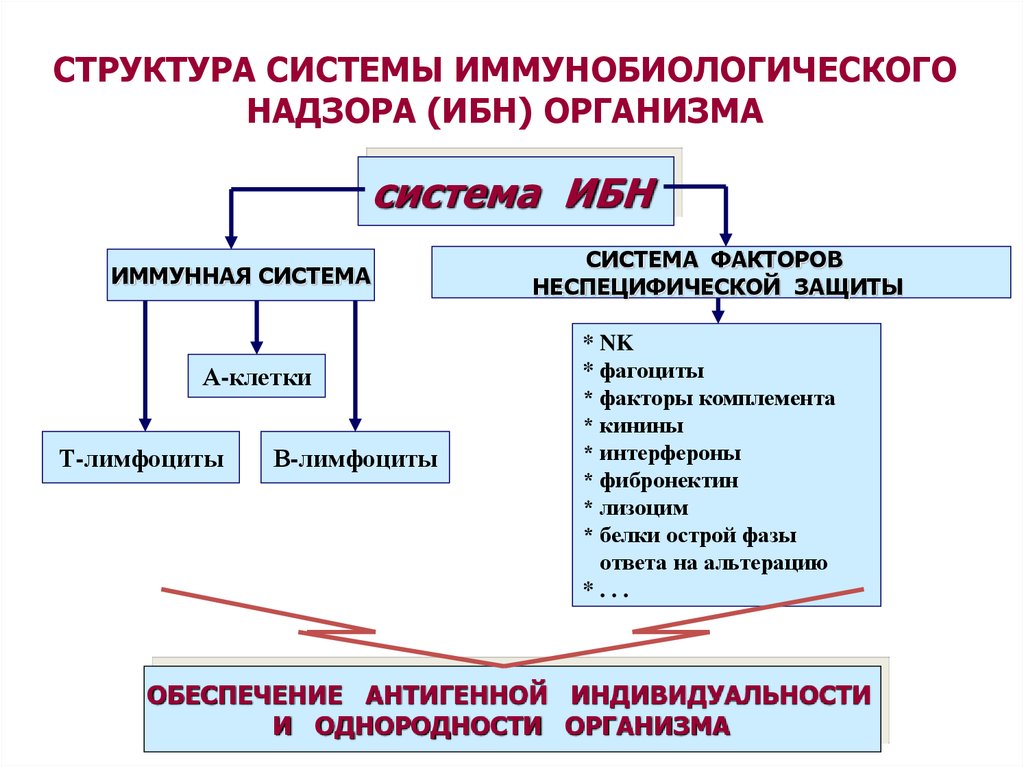
СТРУКТУРА СИСТЕМЫ ИММУНОБИОЛОГИЧЕСКОГОНАДЗОРА (ИБН) ОРГАНИЗМА
система ИБН
ИММУННАЯ СИСТЕМА
А-клетки
Т-лимфоциты
В-лимфоциты
СИСТЕМА ФАКТОРОВ
НЕСПЕЦИФИЧЕСКОЙ ЗАЩИТЫ
* NK
* фагоциты
* факторы комплемента
* кинины
* интерфероны
* фибронектин
* лизоцим
* белки острой фазы
ответа на альтерацию
*...
ОБЕСПЕЧЕНИЕ АНТИГЕННОЙ ИНДИВИДУАЛЬНОСТИ
И ОДНОРОДНОСТИ ОРГАНИЗМА
5. Специфическая и неспецифическая иммунная защита
• Неспецифические факторы иммунитета, такие какфагоциты, естественные киллерные клетки и
комплемент (особые ферменты) могут бороться с
инфекцией как самостоятельно, так и в
кооперации со специфической защитой.
• Под специфической защитой понимаются
специализированные
• лимфоциты, которые
• могут бороться только
• с одним антигеном.
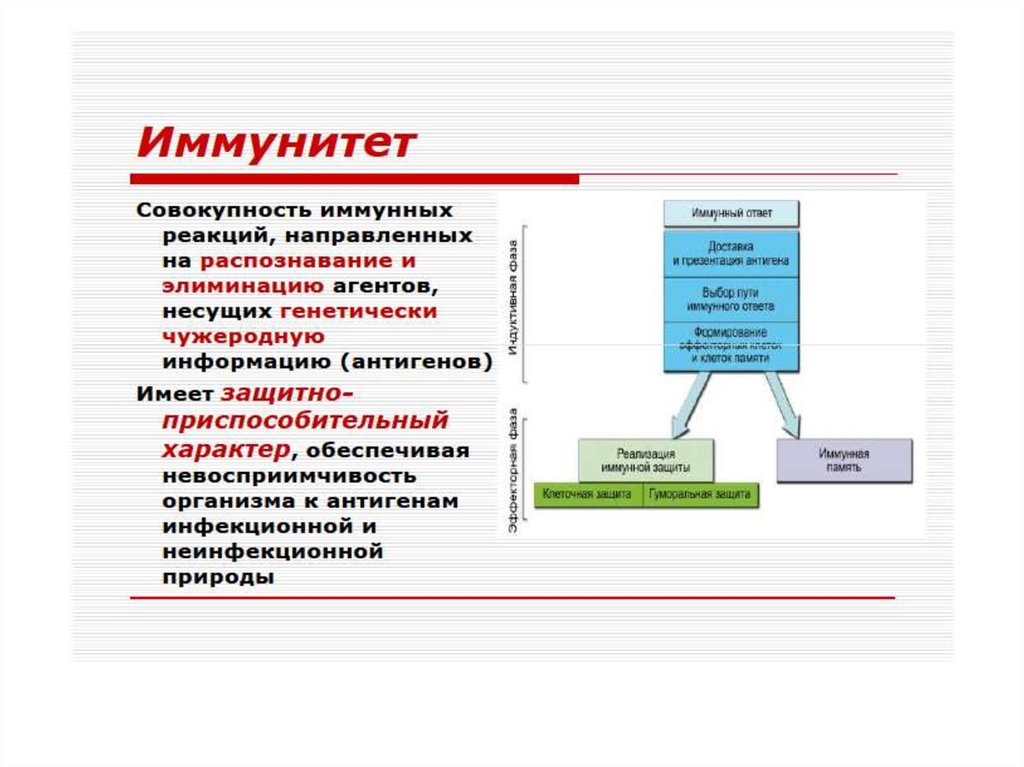
6. И М М У Н И Т Е Т
ИММУНИТЕТ• * Физиологическая форма иммуногенной
реактивности.
• * Формируется в результате реализации наследуемой
генетической программы и/или при контакте клеток
иммунной системы с чужеродным ей антигеном.
• * Обеспечивает постоянный и однородный
антигенный состав организма.
• * Реализуется путем обнаружения, как правило, деструкции,
инактивации и элиминации чужеродного антигена.
• * Характеризуется повышенной резистентностью
организма к нему.
7.
АНТИГЕН(греч. anti - против, genes - порождающий,
создающий, вызывающий)
* вещество экзо- или эндогенного происхождения,
* как правило, вызывающее иммунную реакцию.
8.
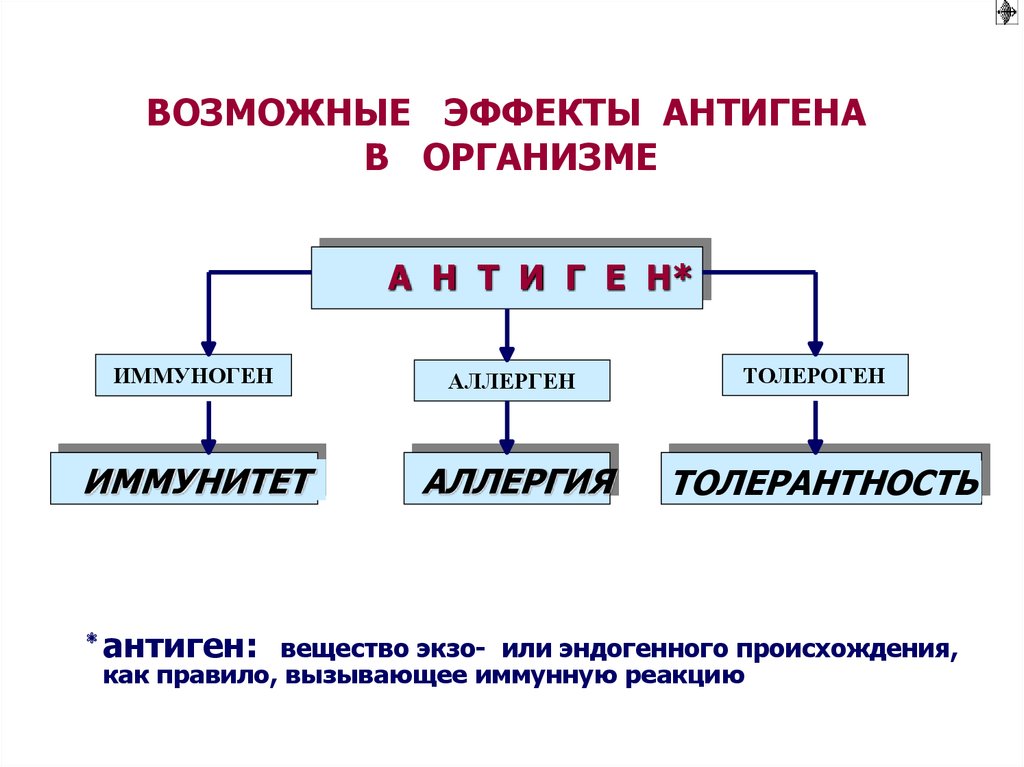
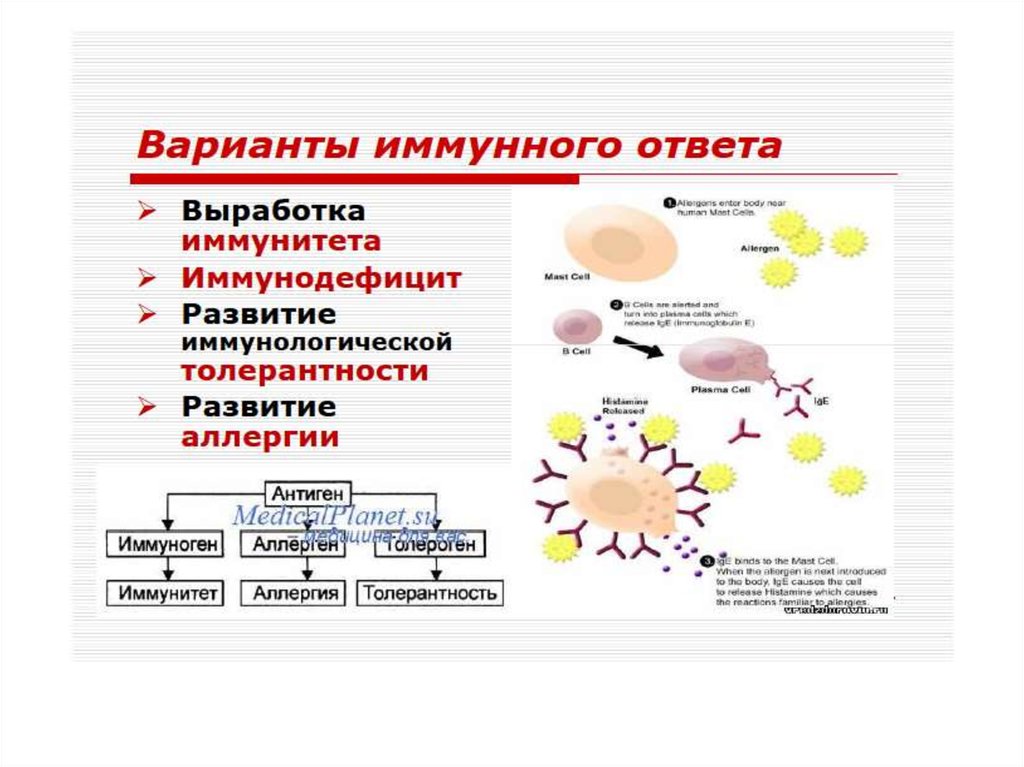
ВОЗМОЖНЫЕ ЭФФЕКТЫ АНТИГЕНАВ ОРГАНИЗМЕ
А Н Т И Г Е Н*
ИММУНОГЕН
АЛЛЕРГЕН
ИММУНИТЕТ
АЛЛЕРГИЯ
ТОЛЕРОГЕН
ТОЛЕРАНТНОСТЬ
* антиген: вещество экзо- или эндогенного происхождения,
как правило, вызывающее иммунную реакцию
9.
Антитела - это иммуноглобулины, связывающиеся с поверхностнымиантигенами микроорганизмов и с продуктами их жизнедеятельности
(например, с токсинами).
IgA - препятствует адгезии и проникновению микроорганизмов и токсинов в
подслизистую и систему кровообращения. Связанные с IgA микробы и токсины
удаляются за счет механического клиренса. IgA реакция на инфекцию не
сопровождается болезнью.
IgG связываются с микробами и токсинами в глубине тканей или в
циркуляторном русле. Они активируют систему комплемента и фагоциты
(нейтрофилы), таким образом, уничтожая бактерии и токсины. IgG ответ
сопровождается болезнью.
IgE связываются с базофилами и тучными клетками. При встрече с паразитом
они вызывают выброс гистамина и других биологически активных веществ из
этих клеток. Гистамин привлекает эозинофилы, которые оказывают
повреждающее действие на паразитов. При нарушенном иммунном ответе, IgE
может вырабатываться против непаразитарных антигенов, что приводит к
аллергии.
10.
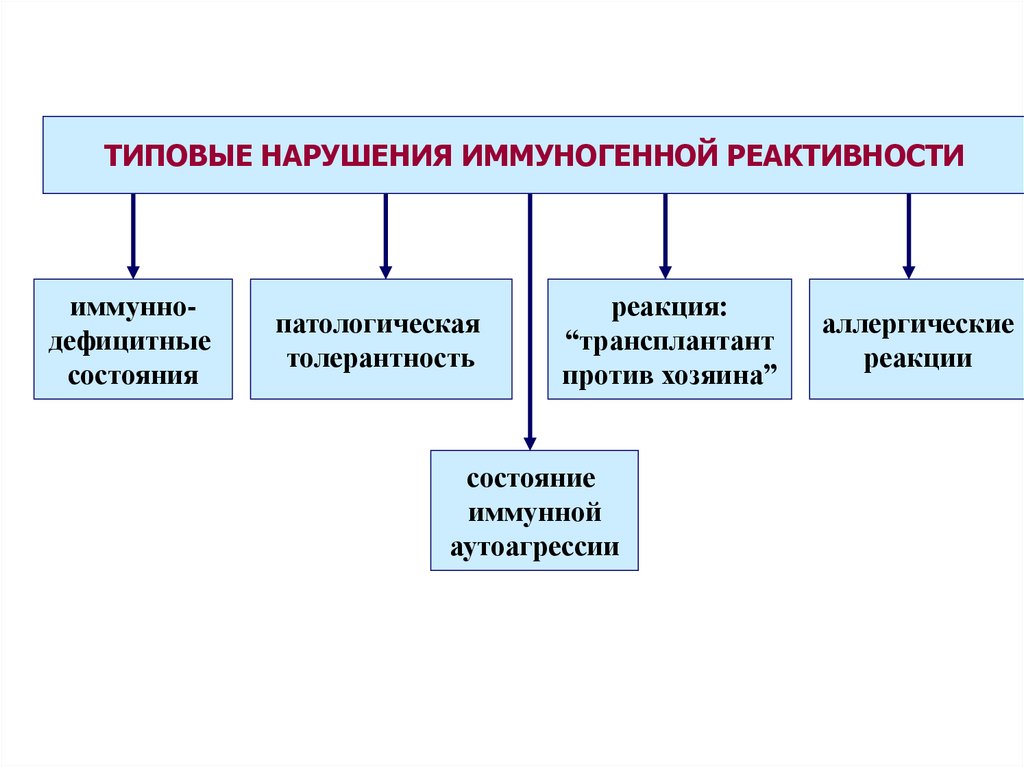
ТИПОВЫЕ НАРУШЕНИЯ ИММУНОГЕННОЙ РЕАКТИВНОСТИиммуннодефицитные
состояния
патологическая
толерантность
реакция:
“трансплантант
против хозяина”
состояние
иммунной
аутоагрессии
аллергические
реакции
11.
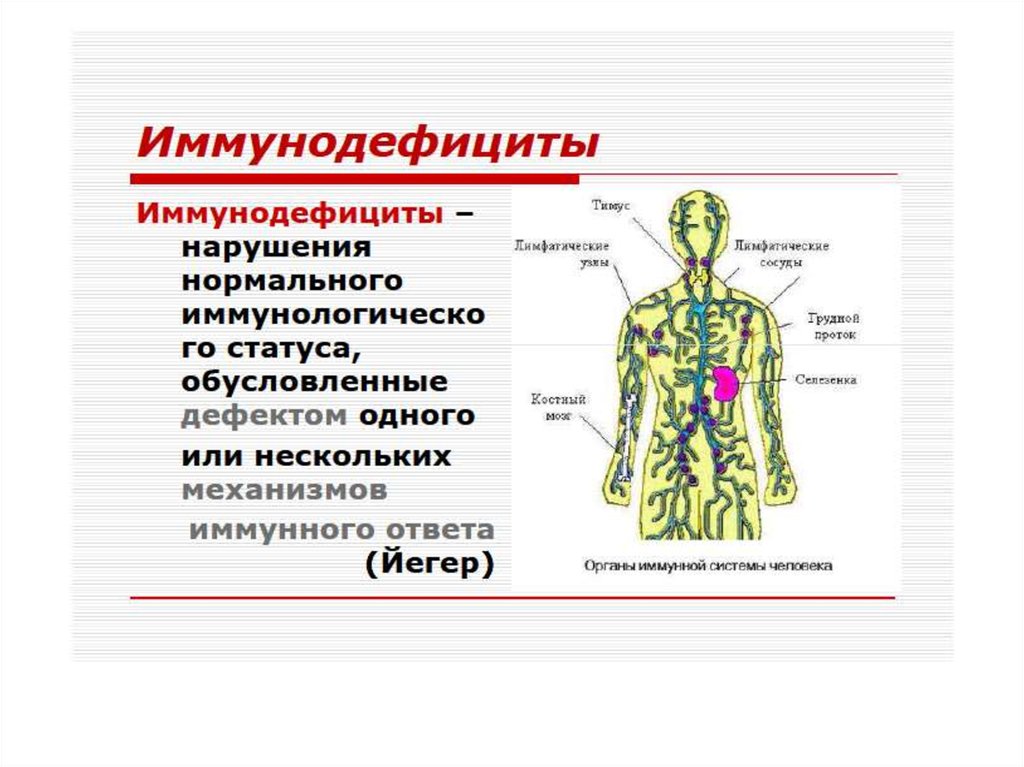
ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ(ИДС)
* Типовая форма патологии
системы иммуно-биологического надзора.
* Характеризуется
снижением эффективности или полной
неспособностью её осуществлять реакции
обнаружения, деструкции и элиминации
чужеродного антигена.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
ТИПОВЫЕ НАРУШЕНИЯ ИММУНОГЕННОЙ РЕАКТИВНОСТИиммуннодефицитные
состояния
патологическая
толерантность
реакция:
“трансплантант
против хозяина”
состояние
иммунной
аутоагрессии
аллергические
реакции
51.
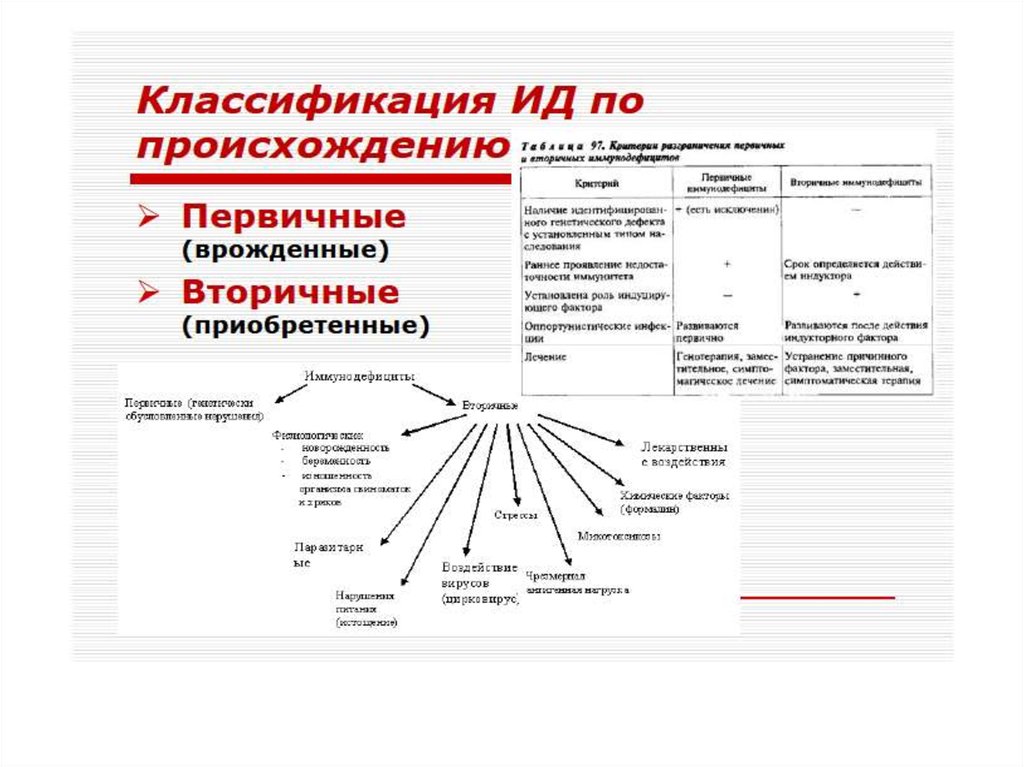
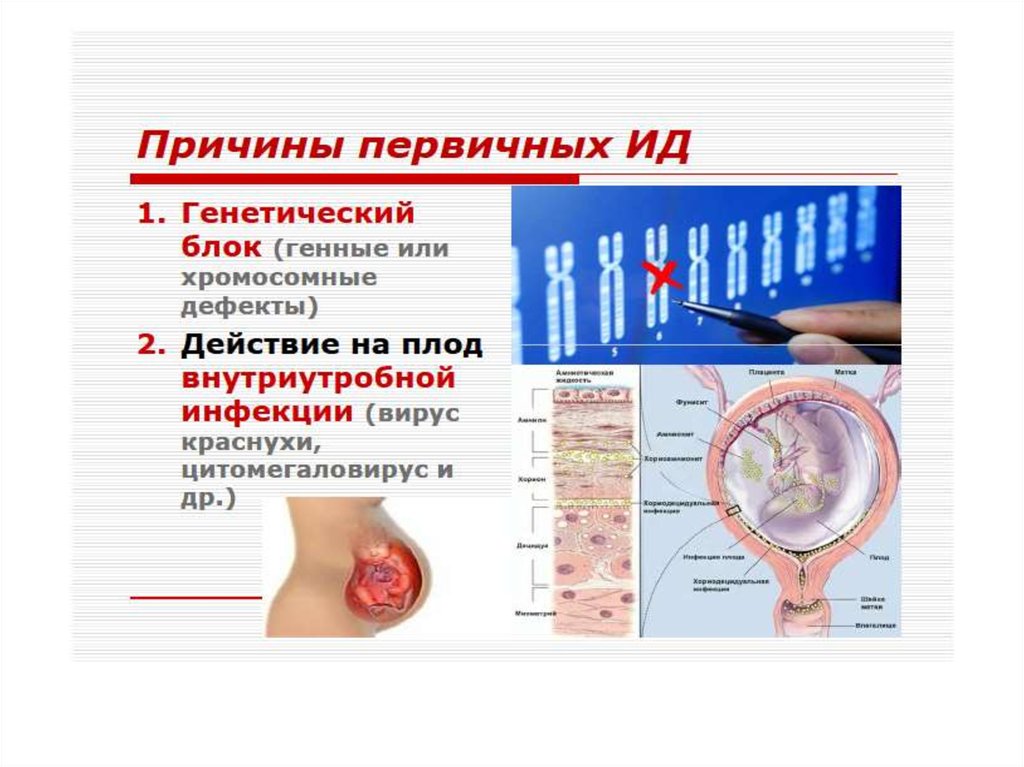
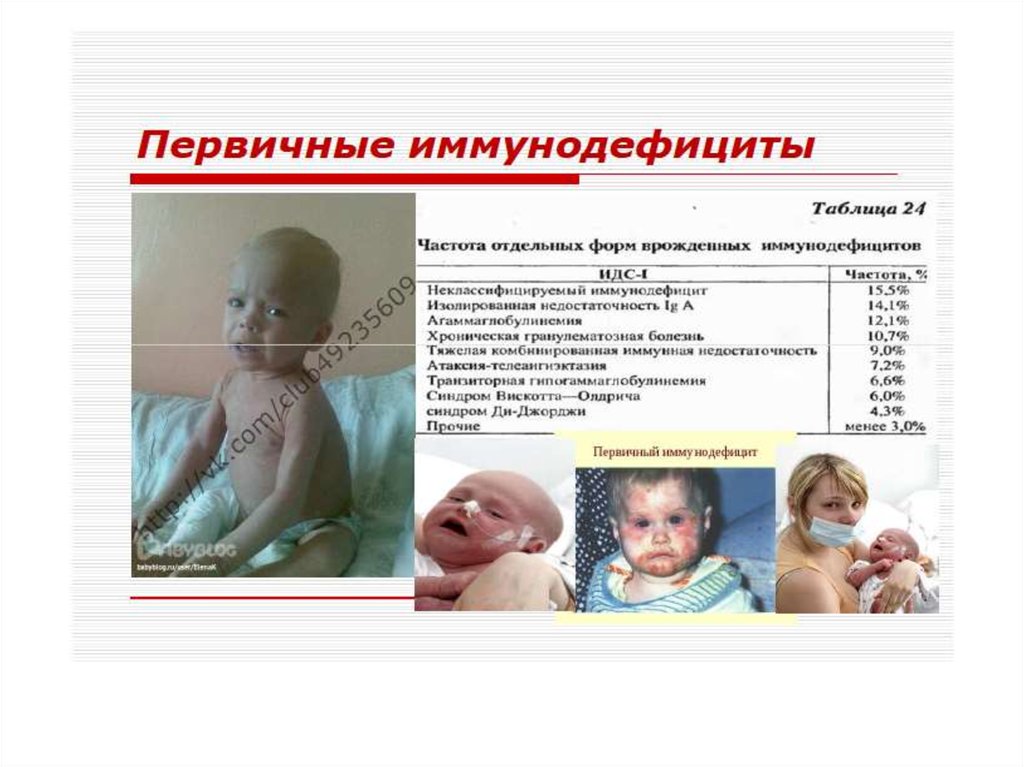
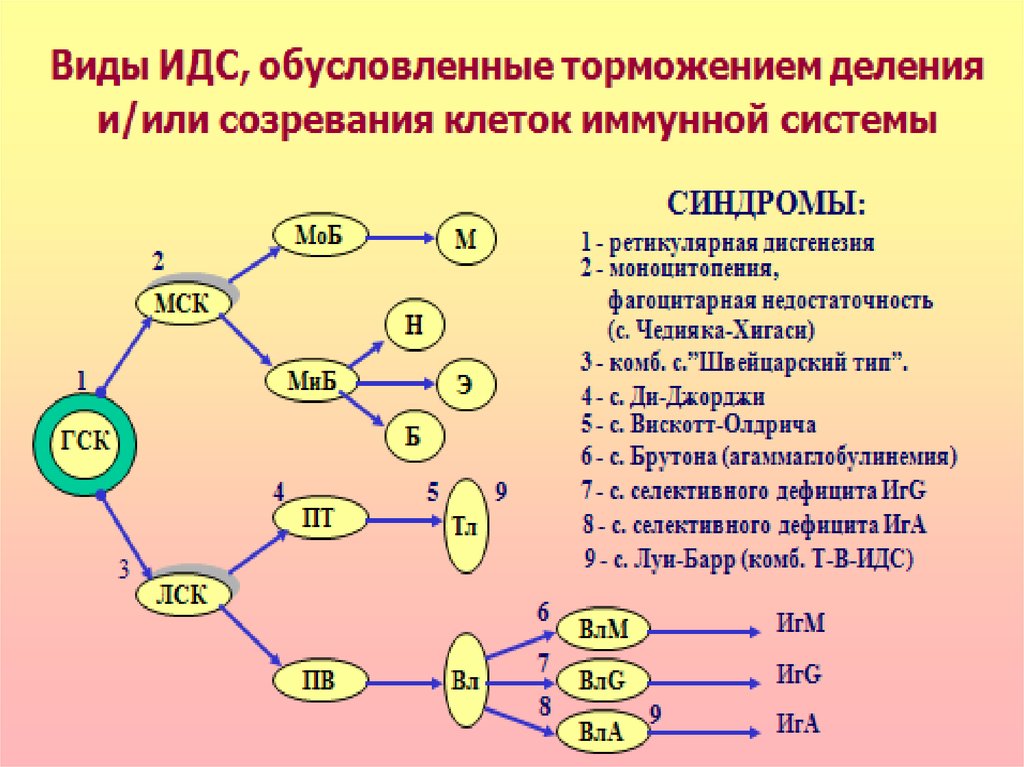
Различают первичные(наследственные) и вторичные
(приобретенные) иммунодефициты.
В основе патогенеза первичных лежит, как
правило, генетически обусловленный блок
развития или выпадение иммунных
процессов вследствие дефекта ферментов
или мембранных структур.
52.
Согласно классификации, предложенной ВОЗ, взависимости от преобладающего типа
поражений звеньев иммунной системы,
различают следующие:
1.комбинированные иммунодефициты с
повреждением гуморального (В) и клеточного (Т)
звеньев иммунной системы
2. с преимущественным повреждением клеточного (Т)
звена иммунной системы
3. с преимущественным повреждением гуморального
(В) звена иммунной системы
53.
Первичные иммунодефицитыПервичные иммунодефициты – очень редкие
состояния (1 на 1 млн. чел). Являются уделом детского
возраста, так как большая часть больных не доживает
до 20 лет, а при более легких формах дефекты со
временем компенсируются.
54.
55.
Комбинированные иммунодефицитыНаиболее тяжелая разновидность ИДС –
синдром ретикулярной дисгенезии, связанный
с нарушением дифференцировки и
пролиферации гемопоэтической стволовой
клетки. Характеризуется значительным
уменьшением в костном мозге стволовых клеток
и блоком созревания лимфоцитов и моноцитов
всех субсистем. Пациенты погибают, как
правило, вскоре после рождения от инфекций
(часто от сепсиса) или злокачественных
новообразований.
56.
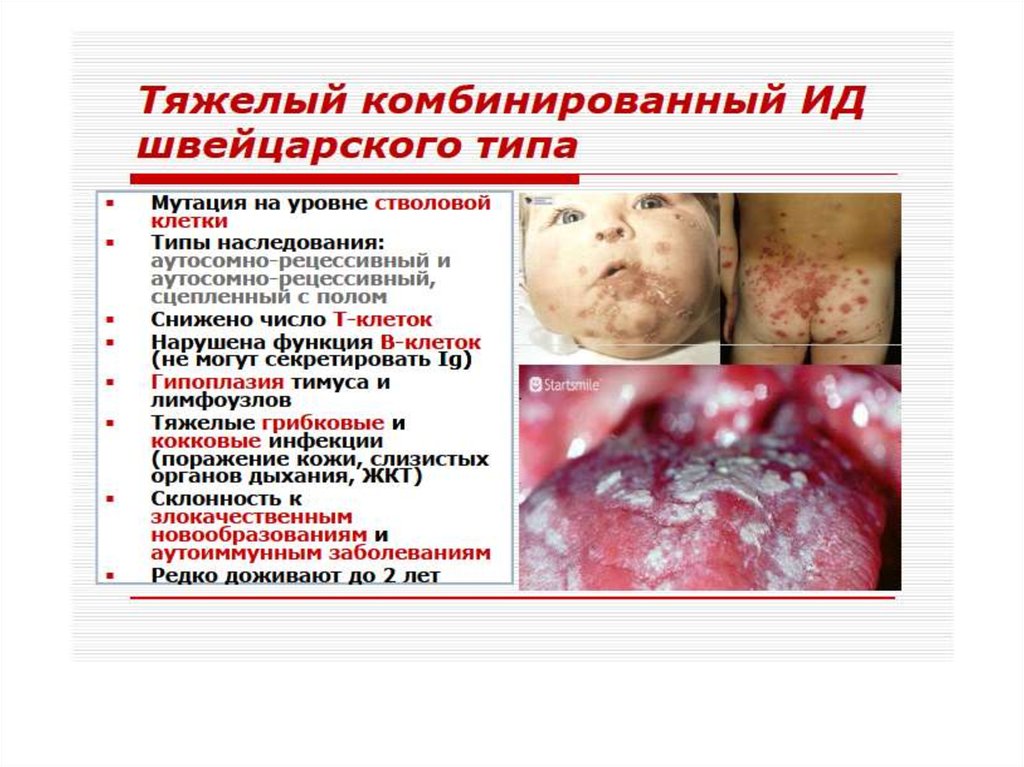
«Швейцарский тип» агаммаглобулинемииразвивается по аутосомно-рецессивному типу.
Дефект развития на уровне лимфоидной
стволовой клетки – нарушение
дифференцировки стволовых клеток, блок
созревания Т- и В-лимфоцитов. Количество Влимфоцитов может быть в пределах нормы, но
они не способны синтезировать
иммуноглобулины (гипогаммаглобулинемия).
Больные погибают в течение первого года жизни
от бактериальных, паразитарных, вирусных,
грибковых инфекций.
57.
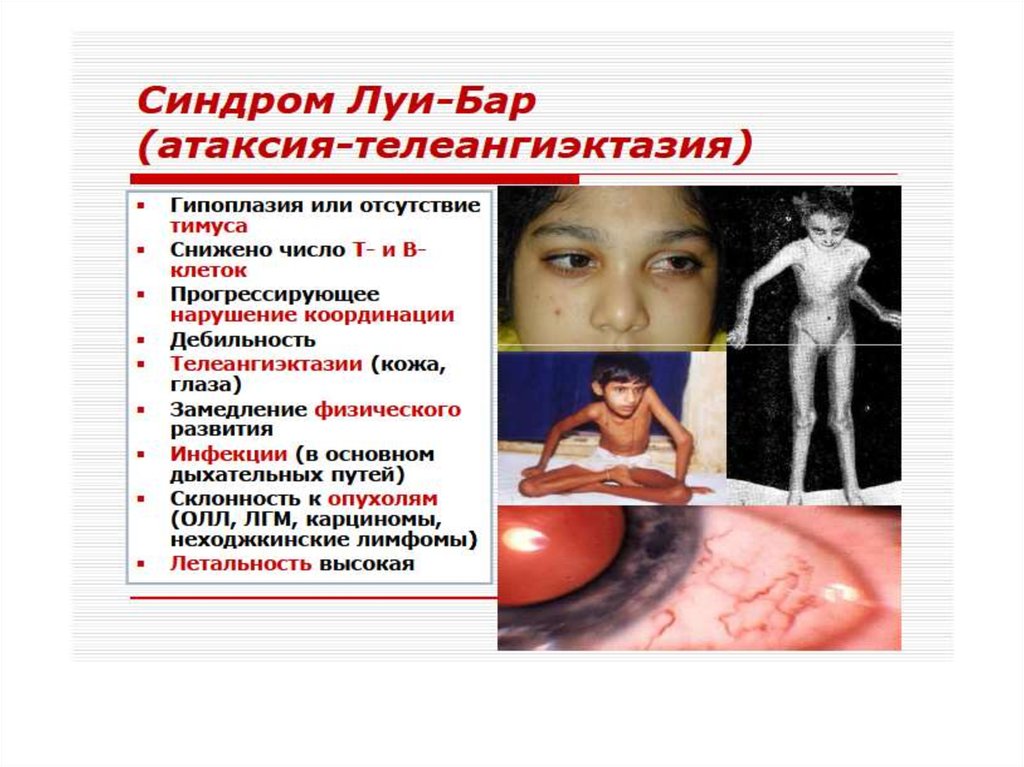
Синдром Луи-Бар описан в 1941 году.Дефект созревания Т- и В-лимфоцитов
(особенно Т-хелперов), резкое снижение их
содержания в крови и дефицит
иммуноглобулинов классов А, Е, реже G.
Пациенты погибают, от инфекций и
опухолей.
58.
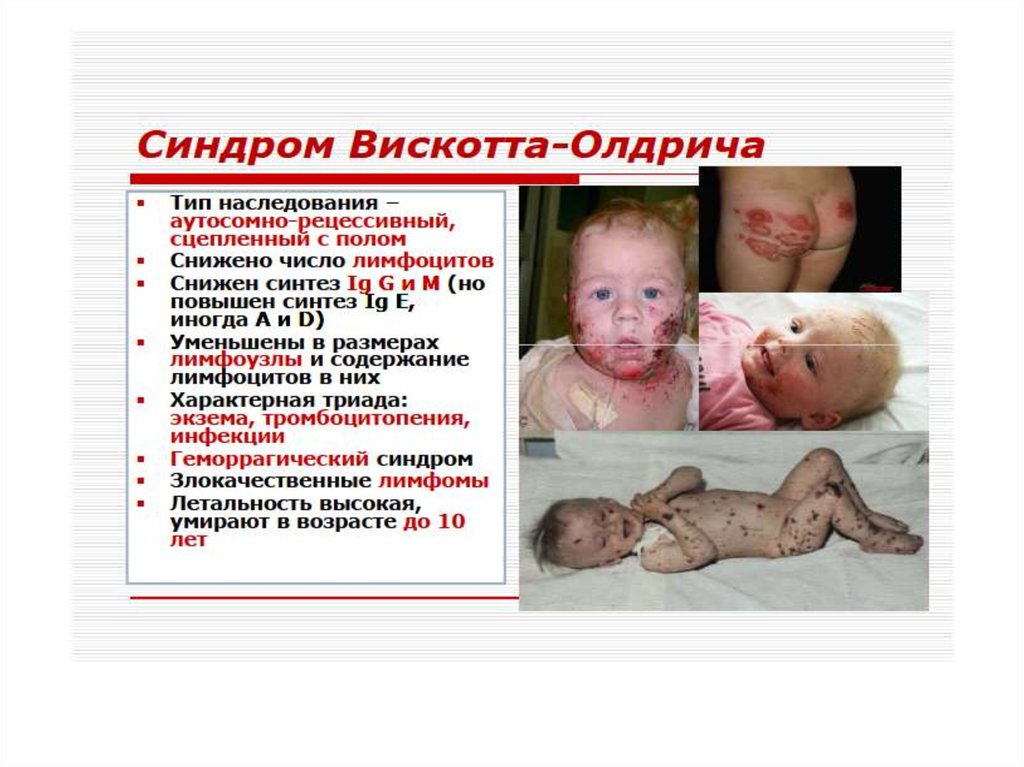
Синдром Вискотта-Олдрича.Нарушение структуры и физико-химических
свойств мембран Т-лимфоцитов. Дефицит
Т-лимфоцитов сочетается с
гипогаммаглобулинемией
дисрегуляторного генеза,
тромбоцитопенией с геморрагическим
синдромом. Частые вирусные и
бактериальные инфекции, аллергические
реакции, экзема.
59.
Атаксия-телеангиэктазия.Комбинированный иммунодефицит:
недоразвитие вилочковой железы,
дефицит Т-клеток и IgG2, IgG4, IgE, IgA.
Неврологическая симптоматика,
поражение сосудистой стенки, нарушение
пигментации. Это обусловлено
нарушением процессов репарации ДНК,
дефектами клеточного цикла
60.
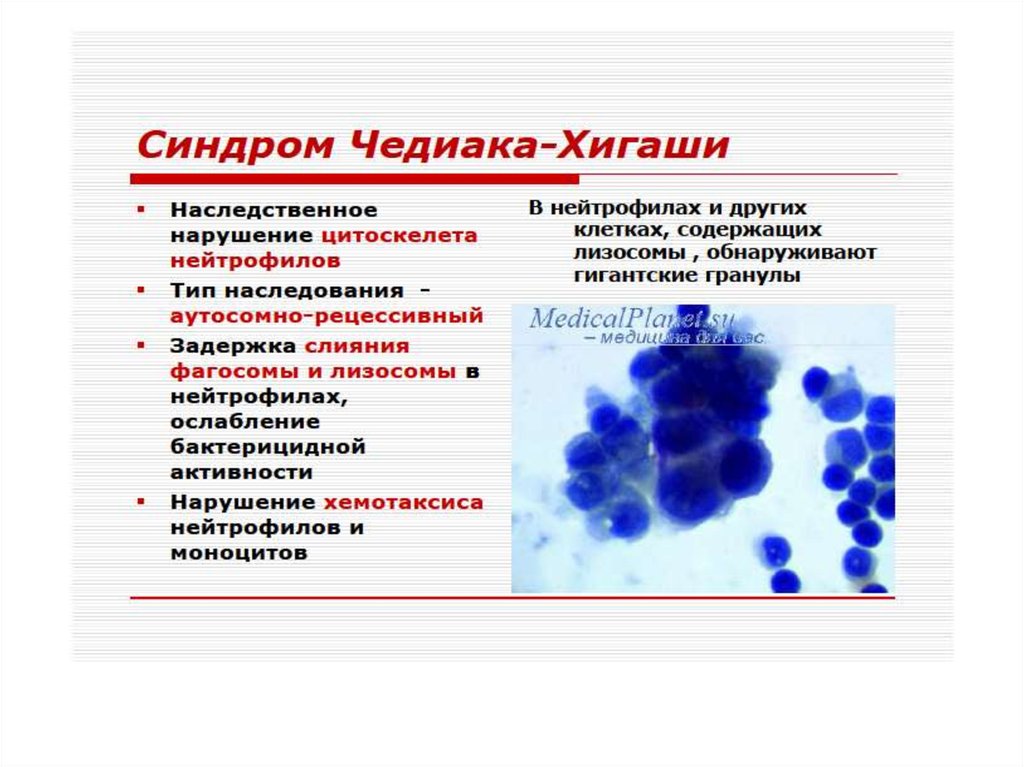
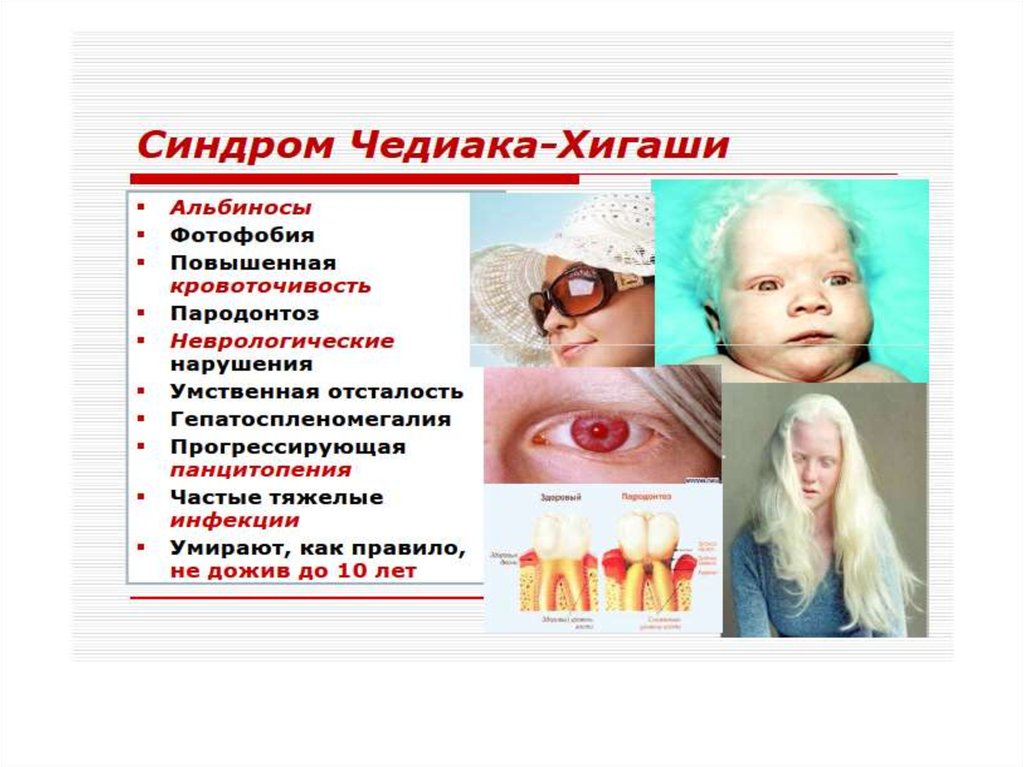
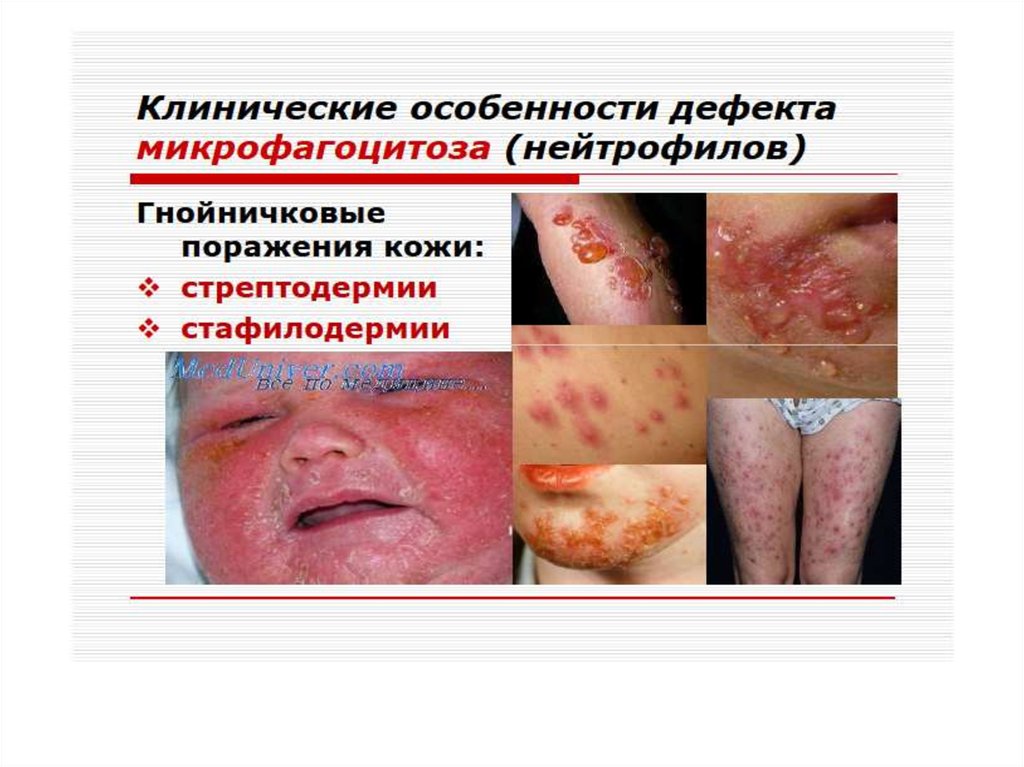
Клеточные иммунодефицитыСиндром Чедиака-Хигаси обусловлен
блокадой пролиферации миелостволовых
клеток, созревания миело- и монобластов.
Сопровождается моноцитопенией,
нейтропенией и фагоцитарной
недостаточностью, что клинически проявляется
частыми хроническими бактериальными
инфекциями.
Нарушения в субсистеме А-клеток (моноциты,
нейтрофилы, эозинофилы, базофилы, т.е. все
клетки, способные к фагоцитозу) проявляются
фагоцитарной недостаточностью
61.
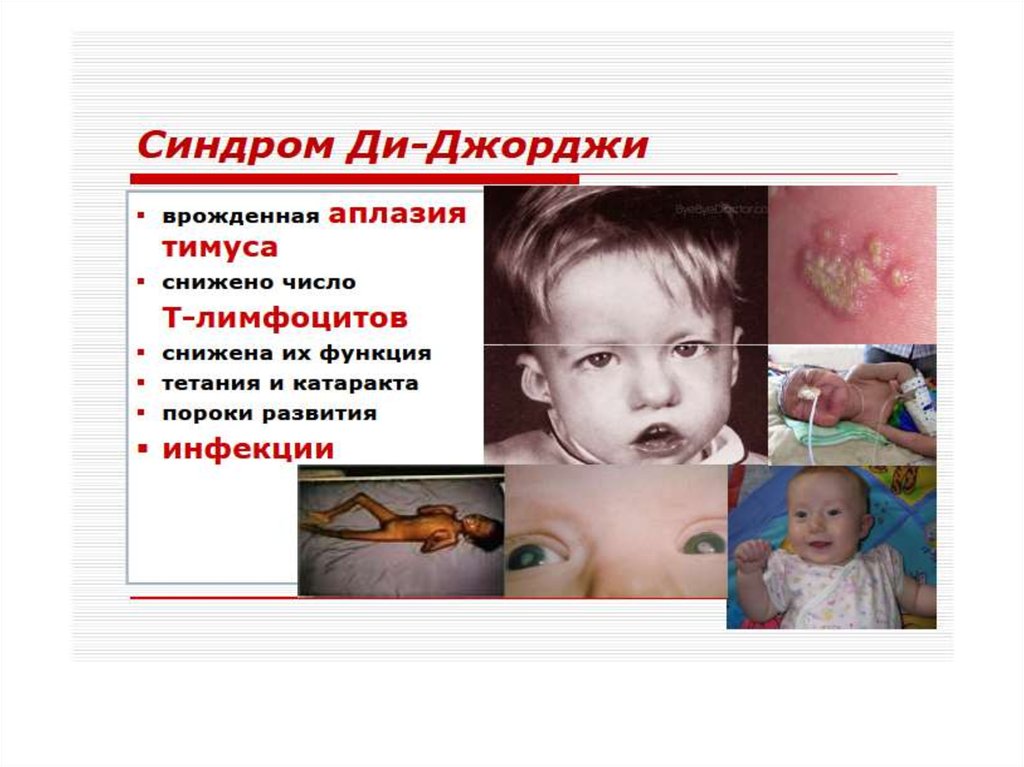
ИД с преимущественным поражением Т-системылимфоцитов. Проявляются развитием инфекций с
внутриклеточным паразитированием возбудителя
(туберкулез, лепра, микозы, вирусные инфекции).
Клиническим проявлением является
синдром Ди-Джорджи. В основе лежит гипоили аплазия стромальных элементов
вилочковой и паращитовидной желез.
Значительный дефицит Т-лимфоцитов. Чаще
болеют девочки. Заболевание характеризуется
гипокальциемией, судорогами,
рецидивирующими инфекциями дыхательных и
мочевыводящих путей.
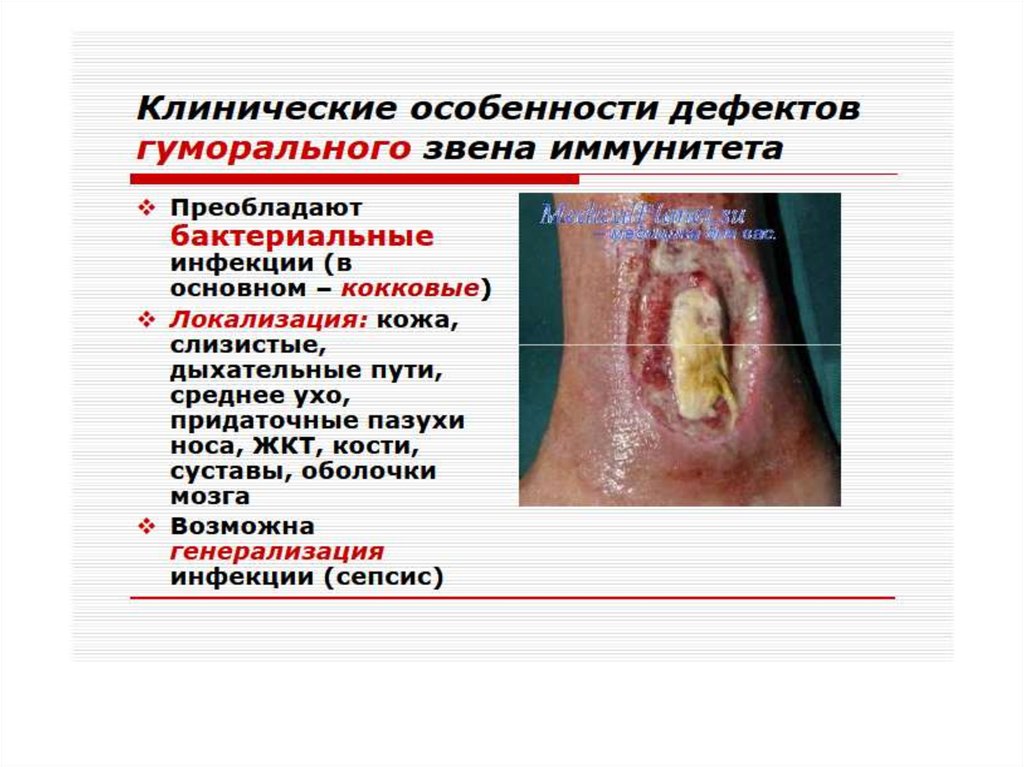
62. Гуморальные иммунодефициты
- характеризуются нормальным уровнем Вл,но пониженной их функциональной
активностью. Клеточный иммунитет
сохранен. У больных отмечается частое
развитие гнойных инфекций
(стрептококовых, пневмококковых),
аутоиммунных процессов, диареи
63.
Синдром Брутона. Описан в 1952 году.Наследственный дефект дифференцировки
В-лимфоцитов, сцепленный с Ххромосомой сопровождающийся гипо- или
агаммаглобулинемией. В основе
патогенеза лежит мутация гена,
контролирующего синтез фермента
тирозинкиназы. Отсутствие этого фермента
нарушает образование
антигенраспознающих рецепторов Вл.
64.
Снижена резистентность организма кстафилококку, стрептококку, пневмококку,
кишечной палочке, сальмонеллам, протею,
вирусным инфекциям, особенно к вирусу
полиомиелита.
Селективные дефициты IgA, IgG –
гипогаммаглобулинемии.
Частые рецидивирующие инфекции
верхних дыхательных путей, желудочнокишечного тракта, конъюнктивы.
65.
Приобретенные (вторичные)иммунодефициты
Они развиваются под влиянием различных
повреждающих факторов физического,
химического или биологического характера. К
развитию гуморального вторичного ИДС часто
приводят заболевания, сопровождающиеся
потерей белков или нарушением их синтеза:
ожоговая болезнь, хроническая почечная и
печеночная недостаточность.
66.
К развитию клеточного вторичного ИДСприводят тяжелые вирусные инфекции, т.к.
вирусы часто имеют тропность к Т-лимфоцитам,
грибковые заболевания.
Вторичные ИДС могут быть следствием действия
повреждающих факторов (радиация,
цитостатики, глюкокортикоиды, антибиотики и
пр.).
В то же время, известны физиологические
иммунодефициты, свойственные раннему
постнатальному и старческому возрастам,
связанные с беременностью.
67.
Физиологический иммунодефицитноворожденных.
Он характеризуется неполноценностью
клеточного и гуморального звеньев ИО, а
также факторов неспецифической
резистентности. Большое количество
лимфоцитов в периферической крови
сочетается со снижением функциональной
активности Т- и В-лимфоцитов. В основном
образуются IgМ, а содержание IgG и IgА
снижено и достигает уровня взрослых
только к 11-14 летнему возрасту.
68.
Отмечается низкая фагоцитарная активность иопсонизирующая способность крови. Уровень
комплемента снижен и нормализуется к 3-6
месяцу жизни.
Иммунный статус беременных женщин
отличается снижением числа и функций Т- и Влимфоцитов, что, по-видимому, связано с
увеличением содержания и активности Тсупрессоров. Это необходимо для подавления
иммунного ответа на антигены плода.
69.
При старении снижается активность какгуморального, так и клеточного звеньев
иммунитета. Снижаются уровни
нормальных антител в крови и способность
к их синтезу. Значительно уменьшается
выработка антител IgG, IgА, а также
активность неспецифических механизмов
защиты. Угнетается синтез IgЕ, поэтому
аллергические реакции протекают не так
остро. С возрастом учащаются и становятся
более активными аутоиммунные болезни
70.
Синдром приобретенногоиммунодефицита
СПИД – тяжелое, с летальным исходом
заболевание вирусной этиологии, которое
поражает иммунную систему организма человека,
передается при половом контакте, через кровь,
при повреждении кожи и слизистых и проявляется
генерализованной лимфаденопатией,
оппортунистическими инфекциями
(протозойными, грибковыми, бактериальными,
вирусными) и онкогенными поражениями.
71.
Первые случаи заболевания былизарегистрированы в 1981 г. в США.
Наиболее высокая инфицированность
наблюдается в странах Центральной и
Восточной Африки (от 4 до 12-20%). На
втором месте по распространенности
заболевания находятся страны
американского континента.
72.
Этиология.Возбудитель СПИДа (AIDS - Acquired Immune
Deficiency Syndrome) относится к ретровирусам
подсемейства лентивирусов (ВИЧ - HIV от англ.
Human immunodeficiency virus). ВИЧ способен к
значительной антигенной изменчивости.
Мутационная активность его в 5 раз выше, чем
вируса гриппа. В организм вирус проникает с
кровью и ее дериватами, с клетками при
пересадке тканей, со спермой и слюной через
поврежденную слизистую.
73.
Попав в организм, возбудитель СПИДавнедряется в клетки, богатые рецепторами
CD-4, к которым гликопротеиды вирусной
оболочки имеют высокий аффинитет.
Наиболее богаты этими рецепторами Тхелперы, но вирус может проникать и в
моноциты, клетки глии, нейроны.
74.
Патогенез СПИДа.ВИЧ, инкорпорированный в геноме клеток в
форме ДНК провируса, способен стимулировать
транскрипцию РНК вируса с помощью ДНКзависимой РНК-синтетазы клетки. На основе этой
РНК синтезируются белковые компоненты
вируса, которые затем интегрируют с его
нуклеиновой кислотой. После завершения
сборки вирусные частицы отторгаются от клетки,
попадают в жидкие среды организма и атакуют
новые клетки, имеющие рецепторы CD-4,
приводя к их гибели.
75.
76.
77.
Массовая гибель Т-хелперов происходит всвязи с взаимодействием вирусного белка
на поверхности зараженной клетки с
рецептором CD-4 на поверхности
незараженных клеток. Одна зараженная
клетка может присоединить к себе до 500
незараженных. Образуется гигантская
нежизнеспособная структура.
Инфицированные клетки впоследствии
лизируются.
78.
Существует несколько версий о механизмахлизиса клеток, пораженных ВИЧ:
1 - мембраны клеток разрушаются при
отпочковывании вируса от клетки. Вероятность
гибели клетки пропорциональна количеству
рецепторов CD-4 на их поверхности, поэтому
количество Тh значительно уменьшается.
2 - рассматривается возможность встраивания
белков вирусной оболочки в клеточные
мембраны и эти клетки распознаются ИКС как
чужеродные и уничтожаются
79.
Считают, что встраивание ДНК вируса в геном Т-хелпералишает их способности к трансформации, созреванию и
реагированию на стимулы, в том числе на ИЛ-2.
Это вызывает их инактивацию и лизис, особенно Тхелперов. Именно поэтому развивается лимфопения.
Кроме того, снижается количество и функциональная
активность естественных NK. Указанные изменения
создают предрасположенность больных к инфекциям,
опухолям, а также к неспособности к развитию
аллергических реакций клеточного типа.
80.
Клиника (стадии болезни).1. Инкубационный период длится от 3-7
нед. до 3-5 лет и более, он более короток
при гомосексуальном и парентеральном
путях заражения и у детей, родившихся от
больных матерей. После попадания в
организм здорового человека вирус
начинает интенсивно размножаться.
Антитела к ВИЧ могут быть обнаружены
уже через 4-8 нед. после заражения.
81.
2. Продромальный период.Характеризуется
-перемежающейся или непрерывной
лихорадкой (38-40ºС) неясной этиологии
(проливные поты, снижение физической
активности).
-- немотивированной потерей массы тела
более 10%, непропорционально уровню
питания и физической нагрузке
- перемежающейся или непрерывной диареей
(не всегда)
82.
При этом у больного развиваютсяперсистирующая (постоянная) генерализованная
лимфаденопатия и умеренный иммунодефицит,
который может сохраняться длительное время
(до 10 лет и более) - так называемый период
скрытого течения инфекции.
3. Стадия вторичных заболеваний
характеризуется нарушениями иммунитета
(лимфопения, резкое снижение Т-хелперов,
подавление функции В-лимфоцитов). Организм
поражается прогрессирующими
оппортунистическими инфекциями:
83.
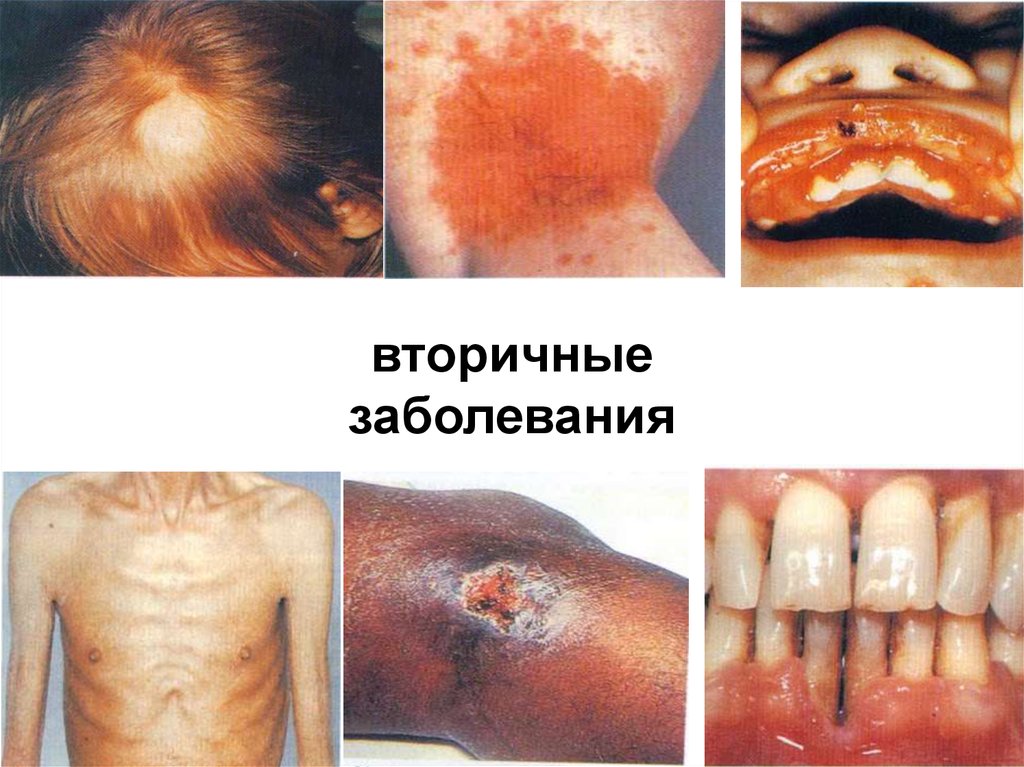
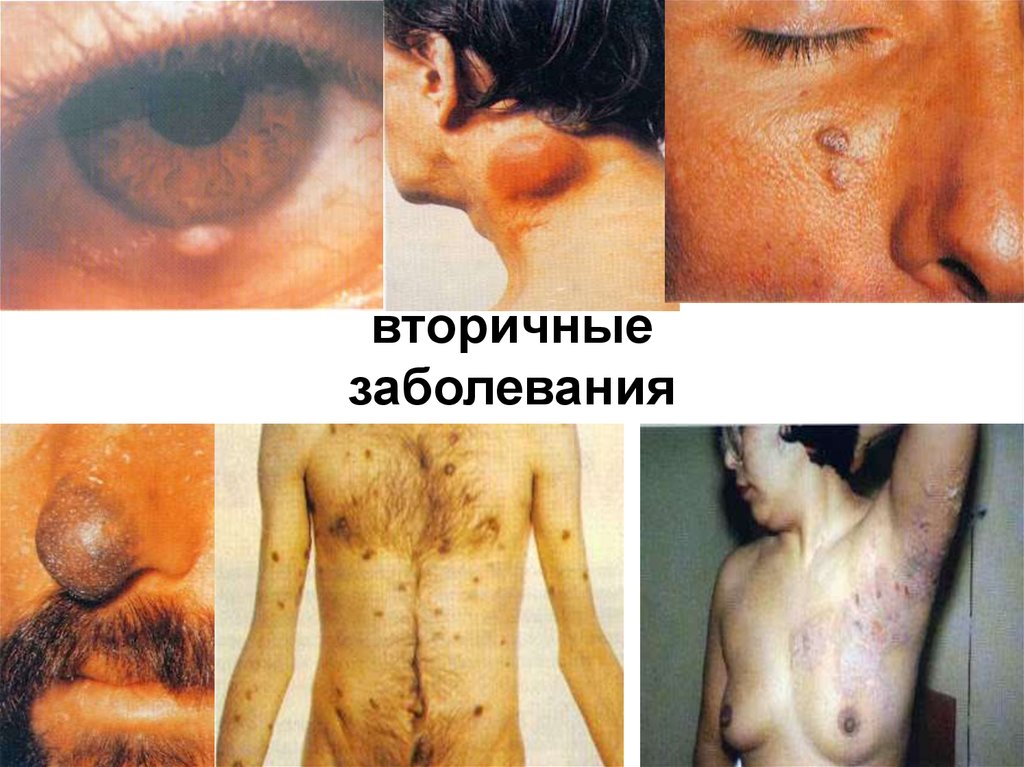
вторичныезаболевания
84.
вторичныезаболевания
85.
повторные и стойкие, часто генерализованныегрибковые, вирусные, бактериальные и
протозойные поражения кожи, слизистых и
внутренних органов,
туберкулез легких и внелегочный,
пневмоцистная пневмония, повторный или
диссеминированный опоясывающий лишай,
кахексия, диссеминированная саркома Капоши,
поражения ЦНС различной этиологии (наиболее
близко стадия Зв соответствует стадии СПИДа по
критериям ВОЗ).
4.Терминальная стадия –заканчивается летально.
86.
Принципы лечения основаны на политерапии:1.Средства, нейтрализующие вирус СПИДа
2.Предотвращение проникновения вируса в
клетки организма
3.Средства, ингибирующие ранние этапы
интеграции ДНК-вируса в геном клетки-хозяина
и др.
87.
Патология неспецифических факторовзащиты
Дефекты системы комплемента.
Комплемент – это система, состоящая из 9
белковых компонентов, являющихся, как
правило, неактивными ферментами и
регуляторными белками. Регуляторные
белки контролируют процессы активации
ферментов, не допуская неконтролируемой
активации комплемента.
88.
Генетический дефект может затронутьлюбой из компонентов как классического,
так и альтернативного путей активации
системы
Иммунологические реакции будут
протекать с отклонениями от нормы,
особенно воспалительные процессы, в
которых система комплемента играет очень
важную роль.
89.
Приобретенная недостаточностькомплемента отмечена при развитии у
человека эндокардита, сепсиса, малярии,
некоторых вирусных инфекций. Все эти
заболевания могут приводить к развитию
гломерулонефрита, возможно, вследствие
накопления неразрушенных в отсутствие
комплемента комплексов АГ-АТ. Активность
комплемента снижается также при СКВ,
ревматоидном артрите и некоторых других
заболеваниях.
90.
Врожденный дефицит фактора С1 – наиболеечастая патология системы комплемента,
наследуется по аутосомному признаку.
Невозможна активация комплемента по
классическому пути, нарушается фагоцитоз и
лизис микробов, наблюдаются повторные и
тяжелые гнойные процессы. При врожденном
дефиците ингибитора С1 (эстеразы) облегчается
активация комплемента и развивается
ангионевротический отек Квинке.
Резистентность к инфекциям при дефиците
ингибитора С1-эстеразы не изменена.
91.
Дефицит С2 – снижение бактерицидности сыворотпредрасположенность к вирусным инфекциям,
диффузным заболеваниям соединительной ткани,
гломерулонефриту и тромбоцитопении.
Компонент С3 является ключевым в формировании
регуляторных и ферментных свойств комплемента
при его дефиците наблюдается высокая смертност
При дефиците ингибитора С3 постоянно активируе
С3, снижается поэтому его содержание в крови.
92.
Нарушаются процессы фагоцитоза и лизисамикробов, снижается сопротивляемость
организма к инфекциям – склонность к
повторным микробно-воспалительным
заболеваниям, лихорадка, кожные сыпи,
артралгии.
При дефиците ингибитора С4 фактора
симптомокомплекс, сходный с системной
красной волчанкой.
93.
Дефицит С5 фактора – нарушенияфагоцитоза и лизиса из-за невозможности
образования соответствующих
компонентов комплемента. Тяжелые
кишечные инфекции, вызванные
грамотрицательной флорой, поражение
кожи. При наследственной
недостаточности С5 с раннего детства
дерматиты и диарея, нарушение развития
ребенка.
94.
Дефицит С6 – заболевания суставов.Дефицит ингибитора С6 – эпизодические
лихорадочные состояния, частые
повторные инфекции
С7 – диффузные заболевания
соединительной ткани: склеродермии,
телеангиэктазии.
С8 – клинически может не проявляться.
95.
Патология пропердиновой системы. Пропердин– белок крови, нормальное антитело,
появляющееся в ответ на иммунизацию
веществами полисахаридной природы.
Соединяясь с полисахаридами микробной
клетки, активирует систему комплемента по
альтернативному пути. Его защитное действие
проявляется в бактерицидном,
вируснейтрализующем эффекте. Снижение
уровня пропердина в крови, а, следовательно, и
его защитных свойств отмечается при тяжелых
патологических процессах (шок, лейкозы,
хронические инфекции).
96.
Патология фагоцитоза.Фагоцитарная недостаточность делится на
следующие виды.
По происхождению
Первичная (наследственная) – большинство
форм передается по аутосомно-рецессивному
типу, реже сцеплено с полом (Х-хромосомой)
Вторичная (приобретенная) – как результат
инфекционно-токсических заболеваний,
нарушений функций печени и почек, системных
болезней соединительной ткани, опухолевой
трансформации мононуклеарных клеток при
миелолейкозе.
97.
По механизму развитияЛейкопеническая.
Приобретенные формы развиваются при
подавлении процессов пролиферации и
созревания моноцитов, в частности при
воздействии радиации, токсинов, цитостатиков.
Например, действие гликолитического яда
монойодацетата, микробных токсинов нарушает
в лейкоцитах выработку АТФ, что приводит к
незавершенному фагоцитозу.
98.
Наследственные формы возникают врезультате наследственной блокады
деления и дифференцировки миелоидной
стволовой клетки.
Дисфункциональная фагоцитарная
недостаточность.
Характеризуется парциальными или
комбинированными расстройствами
различных этапов фагоцитоза и
презентации антигена.
99.
В основе лежат наследственные илиприобретенные дефекты структуры актина
фагоцитов, ферментопатии
глюкозомонофосфатного шунта, гликолиза,
гидролаз лизосом (чаще всего
миелопероксидазы). Дисглобулинемии (чаще с
избытком IgE или недостатком IgG),
сочетающиеся с нарушением процесса
опсонизации и адгезивных свойств фагоцитов.
Недостаточность кислороднезависимых
факторов лизиса объектов фагоцитоза
(лизоцима, лактоферрина, катионных белков).
100.
Дисрегуляторная.Обычно это приобретенная форма,
развивается вследствие нарушения
регуляции различных этапов фагоцитоза
биологически активными веществами.
Нейромедиаторы (катехоламины,
ацетилхолин)
– избыток или недостаток понижают
активность фагоцитоза.
101.
Гормоны.Увеличение концентрации
глюкокортикоидов сопровождается
нарушением процессов деления и
созревания моноцитов, повышением
жесткости их мембран и снижением в связи
с этим их подвижности и адгезивных
свойств.
102.
Лейкокины, простагландины, кинины,пептиды.
Значительное отклонение их концентрации
от нормального диапазона изменяют
характер и интенсивность метаболизма
фагоцитов, состояние их мембран,
активность их ферментов, влияя на
реализацию процессов фагоцитоза, делая
их мало- или неэффективными.