

































 medicine
medicineSimilar presentations:
 Дистрофии. Патоморфология нарушений обмена белков и жиров")
")








Патоморфология нарушений обмена белков, жиров, углеводов (дистрофии)
1.
Патоморфологиянарушений обмена
белков, жиров, углеводов
(дистрофии)
2.
Повреждение (Альтерация)►Повреждение
клеток м. б. летальным
(апоптоз, некроз) и не летальным
(обратимым) в классической
патологии это – дистрофия.
3.
Морфология повреждения►В
физиологических условиях
проявляетс в виде некробиоза и
апаптоза
►В
патологических – в виде
дистрофии и некроза
4.
Дистрофия (dys – нарушение, trophe – питание)– это нарушение метаболизма и структурной
организации клетки (ткани) в процессе
расстройства ферментативных реакций,
сопровождающееся количественным накоплением
в биосистемах продуктов нарушенного обмена с
измененными
физико-химическими свойствами.
5.
Нормальный обмен веществв биосистеме обеспечивается
► Аппаратами
ауторегуляции самой клетки;
► Интегративными системами:
нервной;
гормональной;
транспортной;
иммунной.
6.
Причины дистрофий1.
Нарушения со стороны интегративных и
ауторегуляторной систем (гиперфункционирование
клетки истощение её аварийная стадия
дистрофия).
2.
Ферментопатии (приобретенные и врожденные).
3.
Воздействие токсических и физико-химических
факторов.
4.
Воздействие иммунопатологических механизмов.
7.
Схема патогенеза дистрофий1-е звено – активация лизосомальных ферментов
(стартовая площадка дистрофий).
2-е звено – активация липолиза, протеолиза,
гликогенолиза (накопление недоокисленных продуктов,
изменение рН).
3-е звено – нарушение транспортной системы клетки.
4-е звено – нарушение пластических и энергетических
свойств клетки с накоплением продуктов нарушенного
метаболизма – дистрофия.
8.
Проявления дистрофий► Наиболее
характерные изменения на
клеточном и тканевом уровне
► Накопление
в клетках или между клетками
продуктов нарушенного обмена
► Гистохимическое
обмена
выявление вида продуктов
9.
Морфогенетические механизмыдистрофий
1.
2.
3.
4.
Инфильтрация.
Декомпозиция (фанероз).
Извращенный синтез.
Трансформация.
10.
Инфильтрация- избыточный перенос продуктов обмена из
крови в клетки и ткани и накоплением их
там (пример: СД – почки)
11.
Декомпозиция (фанероз)- поломка мембран ультраструктур клеток с
последующим накоплением продуктов
распада в цитоплазме клеток
с образованием миелиноподобных структур
( в том числе апоптоз)
12.
Извращенный синтез- синтез в клетке веществ, которые в норме
там быть не могут (пример: амилоидоз)
13.
Трансформация- образование продуктов одного вида
обмена из общих исходных продуктов
(появление вместо жиров и углеводов –
белков, при алкогольном гепатите –
«тельца Маллори»)
14.
Классификация дистрофий(4 принципа)
1.
По виду нарушенного обмена (белковая,
жировая, углеводная…).
2.
По происхождению (наследственная,
приобретенная).
3.
По распространенности (местная, системная).
4.
По месту приложения (паренхиматозная,
мезенхимальная, смешанная).
15.
Основные признаки (постулаты)дистрофий
1.
2.
3.
4.
5.
6.
7.
8.
Самый ранний («древний») процесс на раздражение.
Универсальный процесс.
На определенном этапе является охранительным
(приспособительным) процессом.
Служит морфологическим проявлением многочисленных
заболеваний.
Начинается на глубоком (молекулярном) уровне,
затем восходит до клеточного (тканевого).
Развивается на основе физиологических реакций
(количественные превращения переходят в новое качество).
При дистрофии деструкция преобладает над процессами
репарации.
Дистрофия наиболее выражена в высокоспециализированных
органах.
16.
Белковые дистрофии(диспротеинозы)
► Паренхиматозные
(ПД):
гиалиново-капельная;
гидропическая;
роговая;
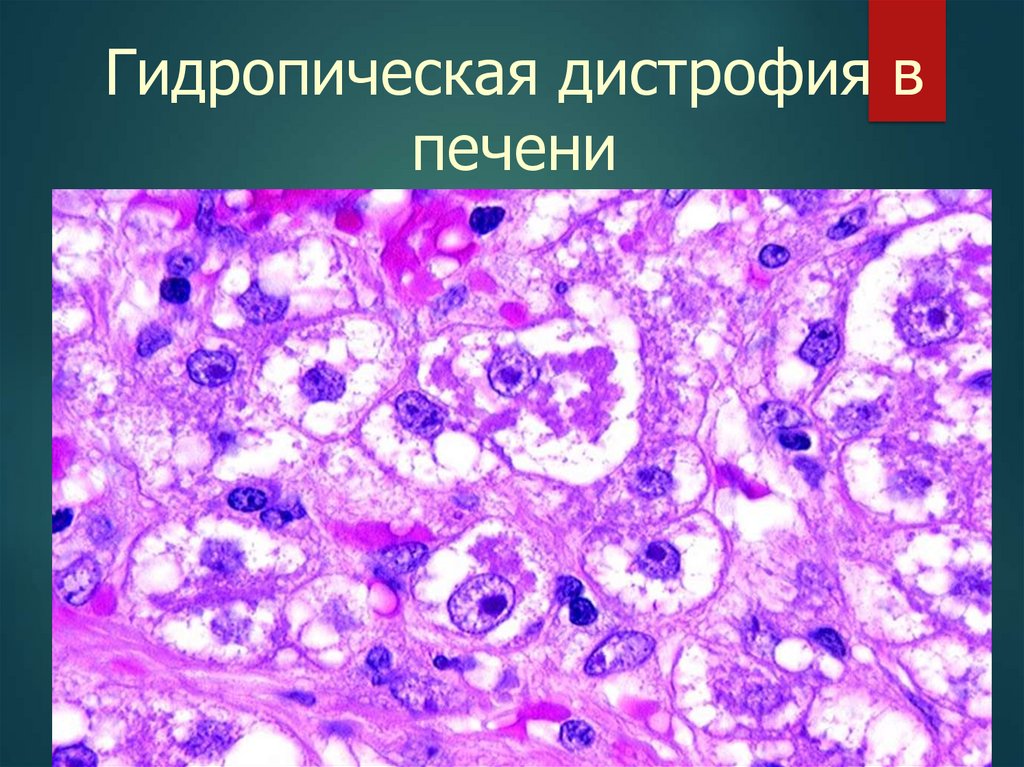
17.
Гидропическая дистрофия впечени
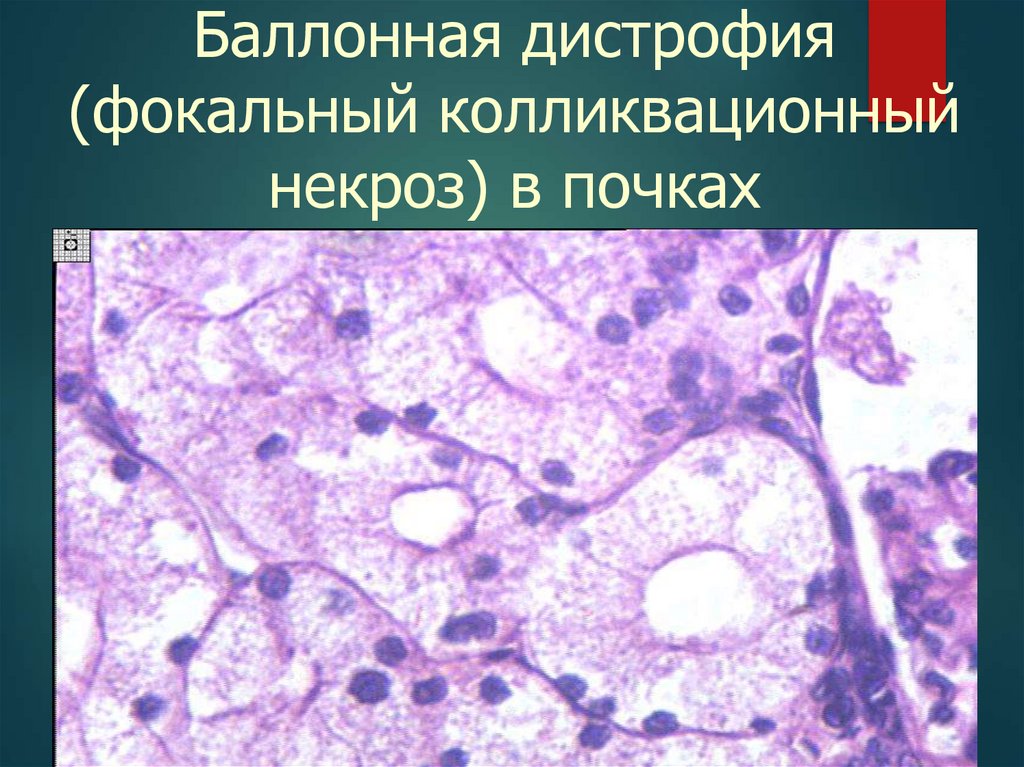
18.
Баллонная дистрофия(фокальный колликвационный
некроз) в почках
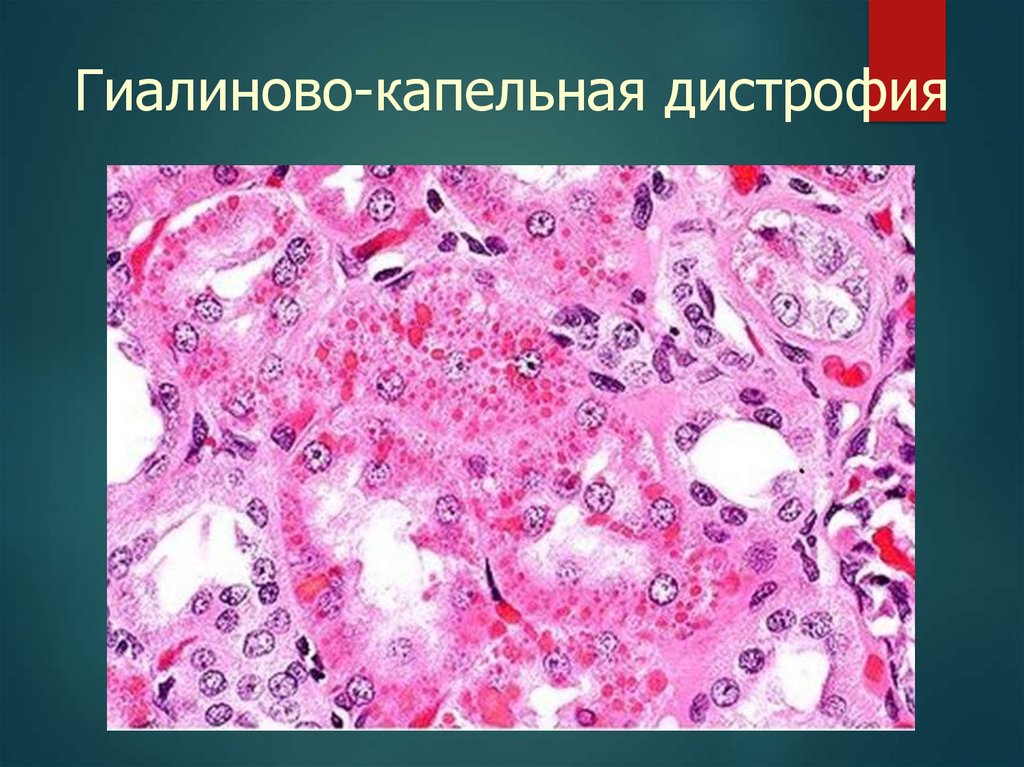
19.
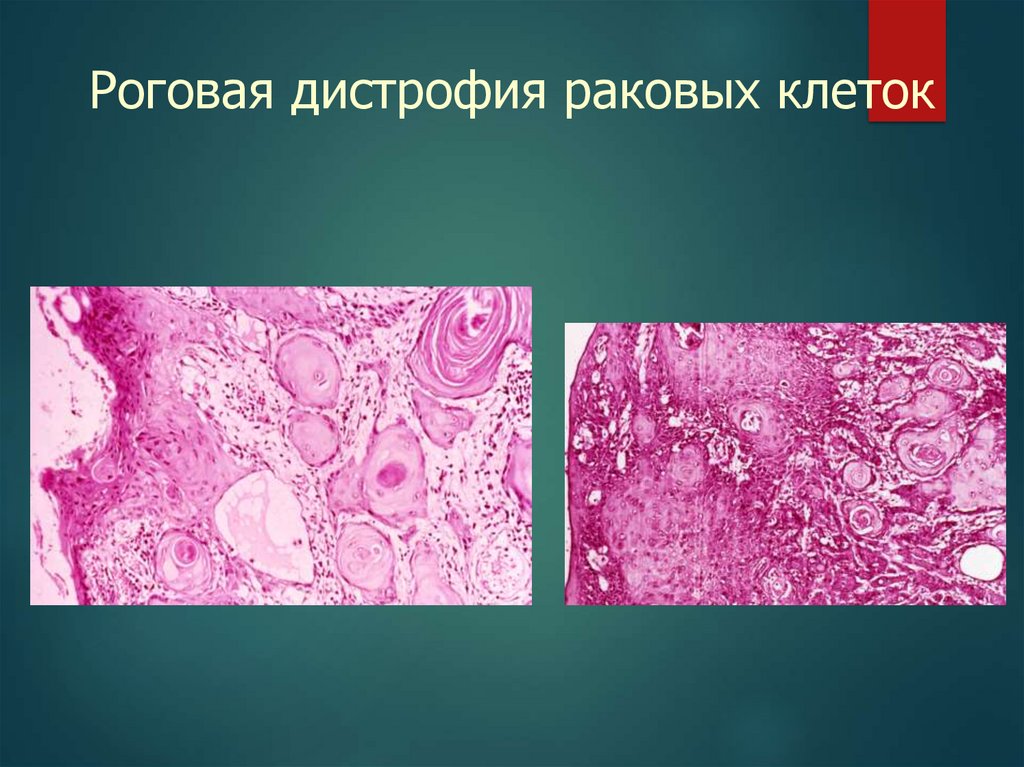
Гиалиново-капельная дистрофия20.
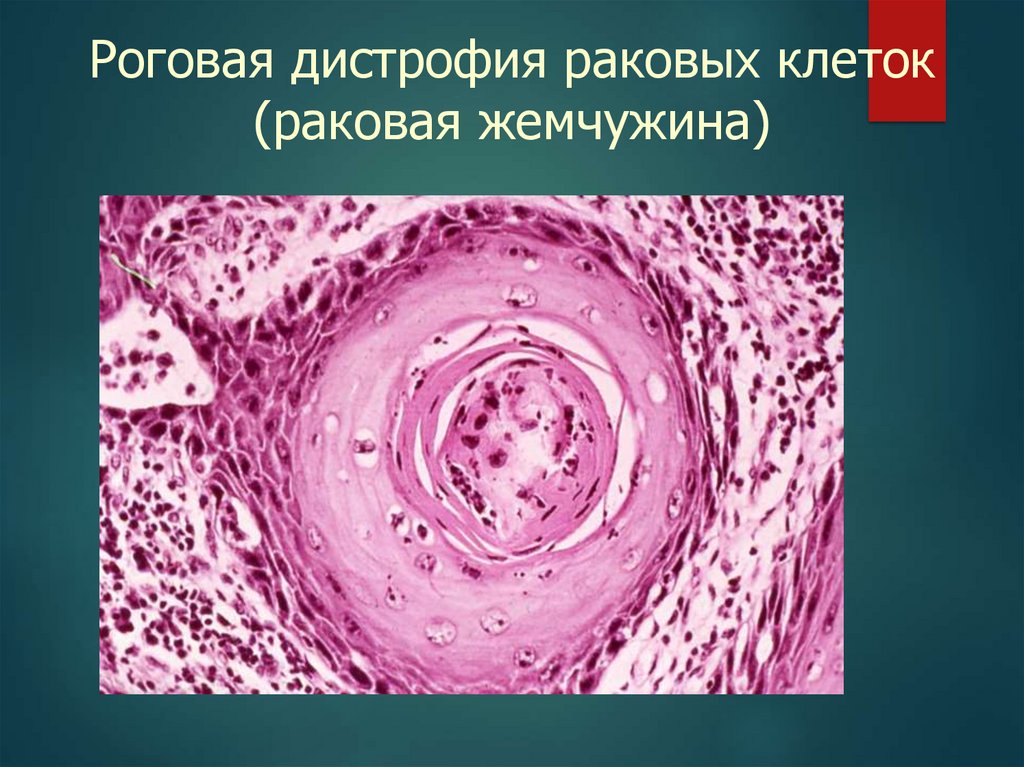
Роговая дистрофия раковых клеток21.
Роговая дистрофия раковых клеток(раковая жемчужина)
22.
Исход паренхиматознойдистрофии
белка гиалиново-капельная
(э/м) гиалиново-капельная (с/м)
коагуляционный некроз (сухая гангрена)
► Коагуляция
белка гидропическая
дистрофия баллонная дистрофия
колликвационный некроз (влажная
гангрена)
► Гидратация
23.
Наследственные ПД1.
2.
3.
4.
Цистиноз.
Тирозиноз.
Фенилкетонурия.
Алкаптонурия и др.
24.
Наследственные дистрофии,связанные с нарушением обмена аминокислот
Название
Дефицит
фермента
Локализация
накоплений
аминокислоты
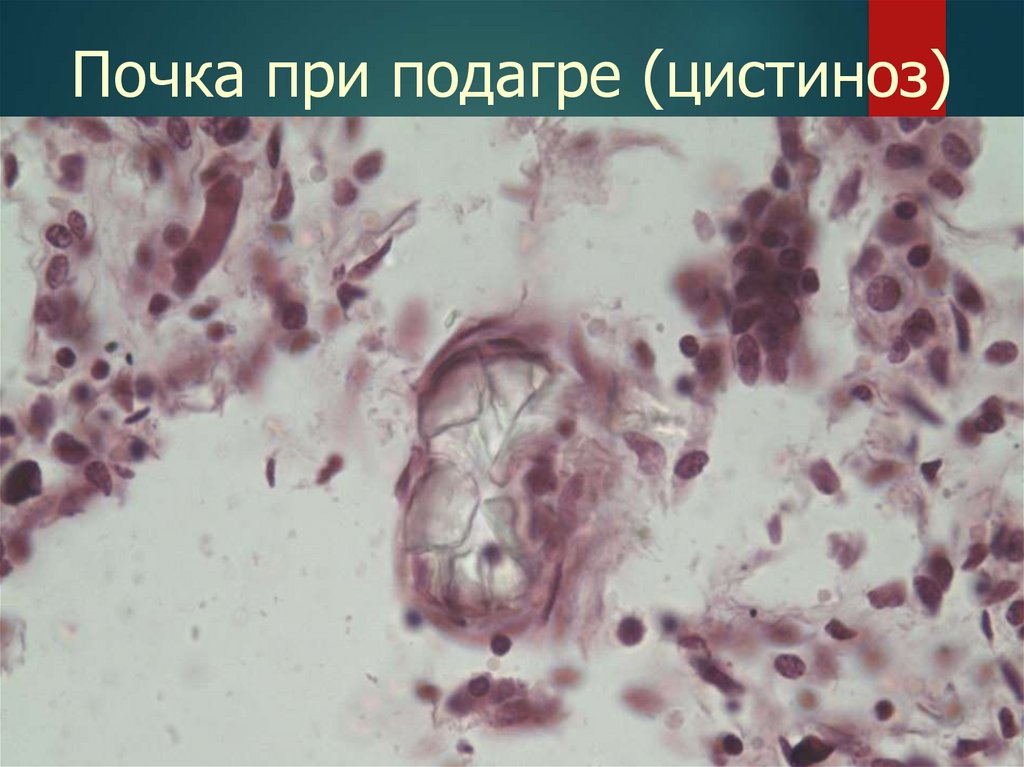
Цистиноз
Неизвестен
Печень, почки,
селезенка, КМ.
ЛУ, кожа
Тирозиноз
Тирозинаминотра
нсфераза
Печень, почки,
кости
Фенилпировиногр Фенилаланин-4адная
гидроксилаза
олигофрения
Нервная система,
мышцы, кожа,
кости
25.
Почка при подагре (цистиноз)26.
Жировые дистрофии (липидозы)► Паренхиматозные
(жировая дистрофия,
наследственный липидоз)
► Мезенхимальные
(ожирение, липоматоз).
27.
Виды жиров1.
2.
3.
4.
Простые (нейтральный жир).
Сложные жиры (фосфатиды,
цереброзиды, ганглиозиды).
Стерины (холестерин).
Стериды.
28.
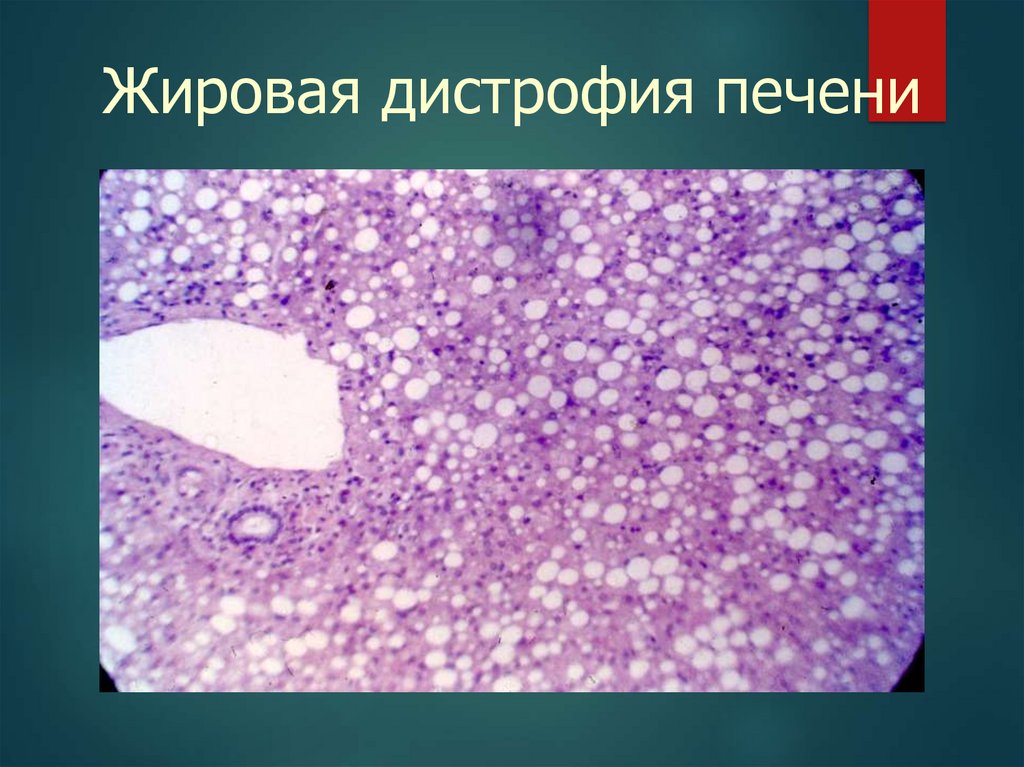
Жировая дистрофия печени(жировой гепатоз) – стадии
1-я – простое ожирение гепатоцитов;
2-я – ЖДП с мезенхимальной реакцией
(воспаление);
3-я – жировой фиброз печени;
4-я – жировой цирроз печени.
29.
Жировая дистрофия печени30.
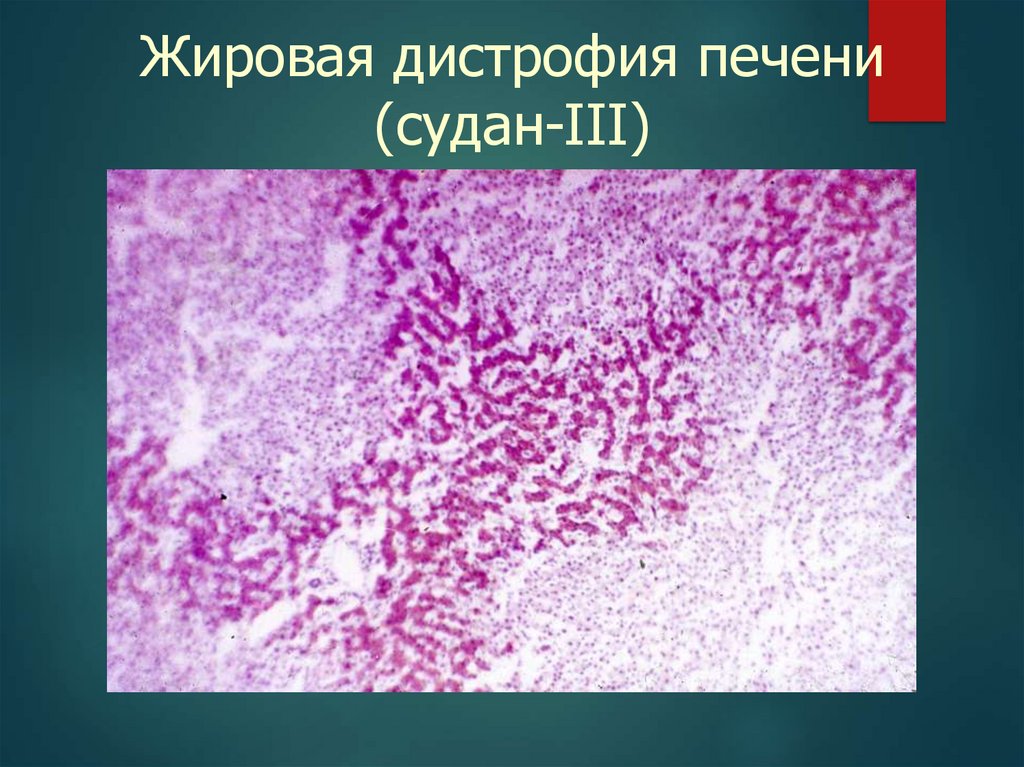
Жировая дистрофия печени(судан-III)
31.
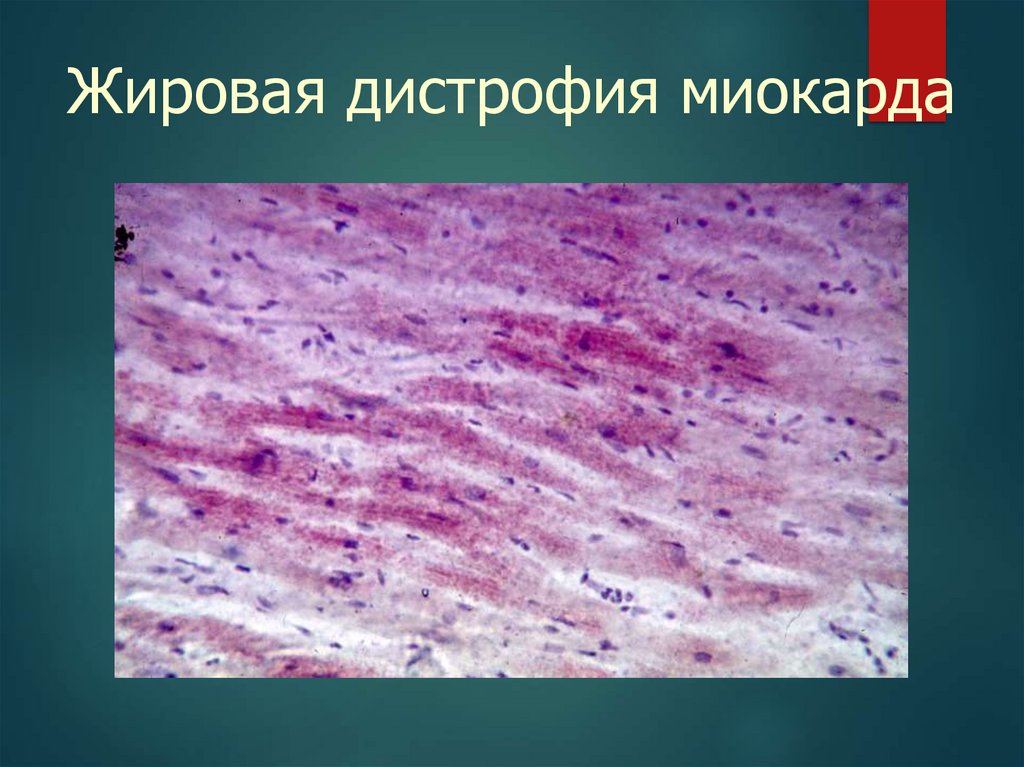
Патогенез жировой дистрофиимиокарда
Гипоксия (дефицит О2) нарушение
окислительного фосфорилирования
анаэробный гликолиз в КМЦ
повреждение митохондрий снижение
АТФ нарушение окисления ЖК
накопление жиров в клетках («тигровое
сердце»).
32.
Жировая дистрофия миокарда33.
Наследственные липидозыВ основе их развития лежит генетический дефект
ферментов. Относятся к болезням накопления –
тезаурисмозам (tesauriso – накапливать). Почти всегда
поражается ЦНС (поэтому говорят о нейролипидозах).
Нейролипидозы подразделяют:
ганглиозидозы (болезнь Тея-Сакса);
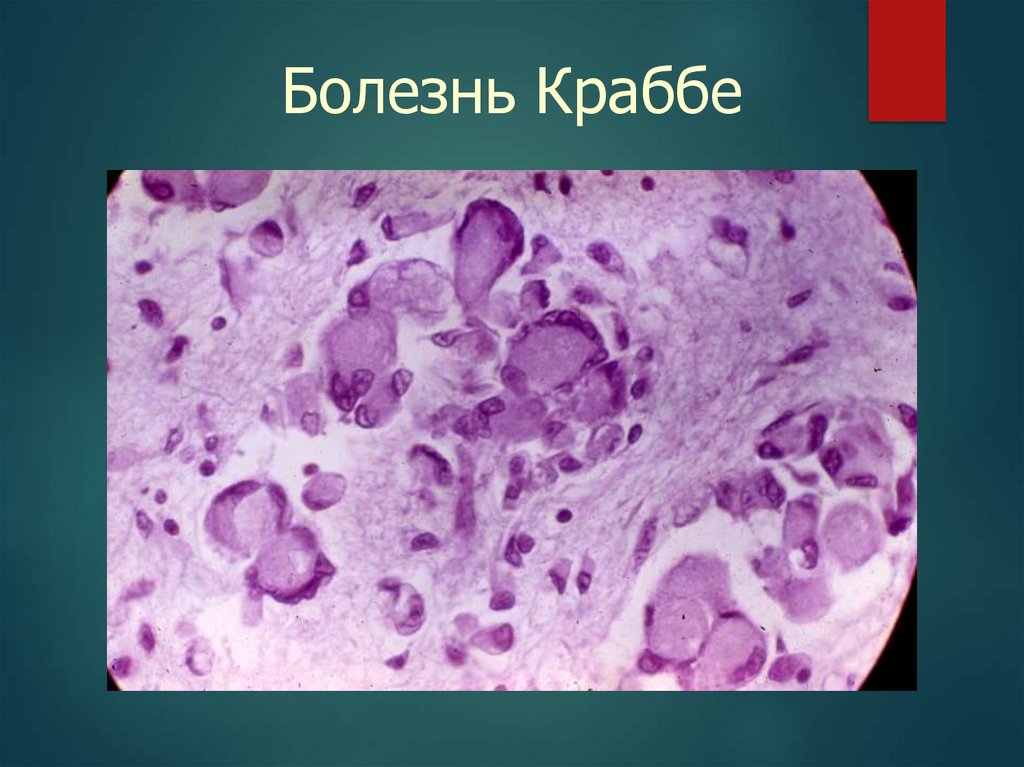
цереброзидозы (болезни Фабри, Краббе, Гоше);
сфингомиелинозы (болезнь Нимана-Пика);
сульфатидозы.
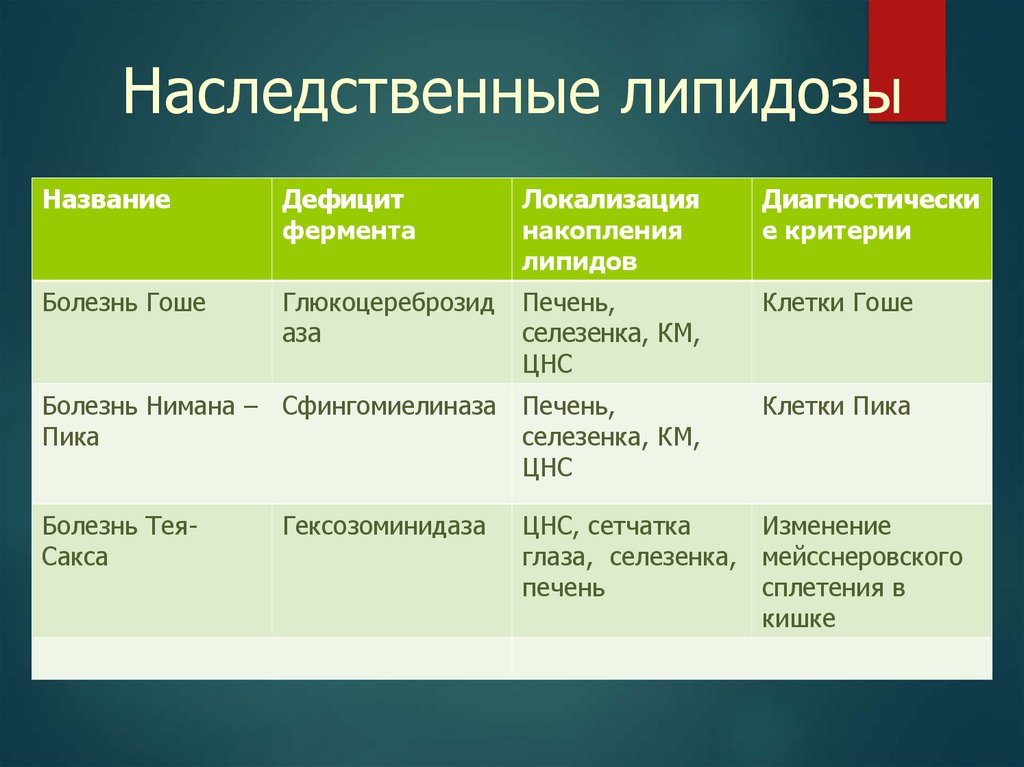
34.
Наследственные липидозыНазвание
Дефицит
фермента
Локализация
накопления
липидов
Диагностически
е критерии
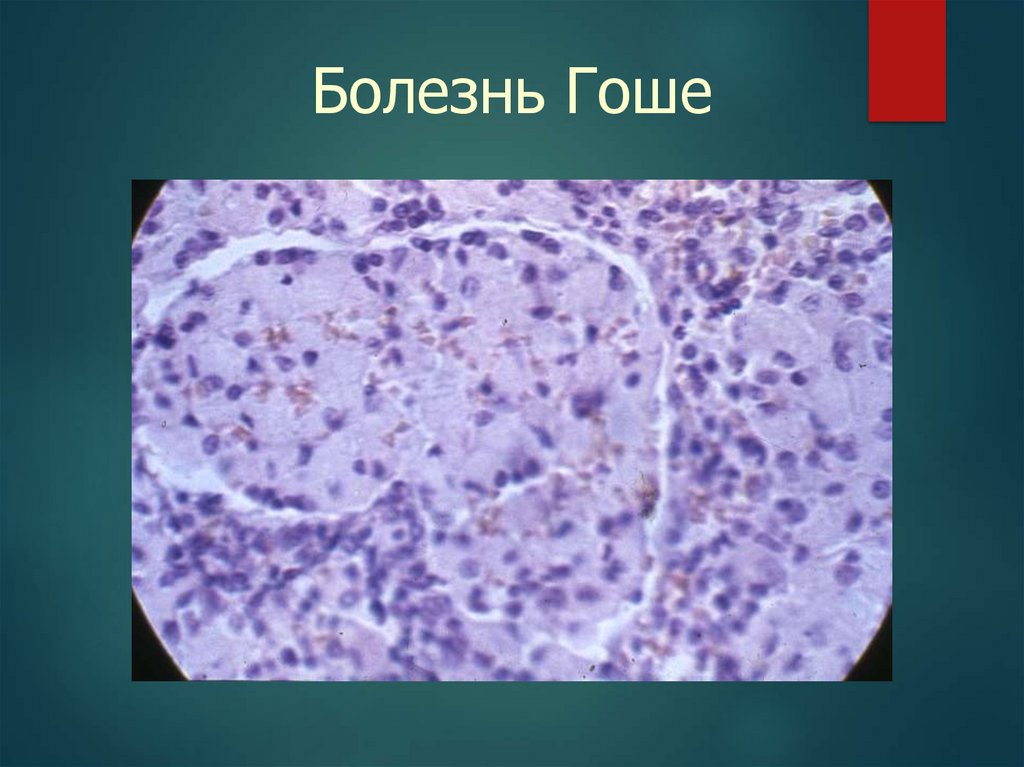
Болезнь Гоше
Глюкоцереброзид
аза
Печень,
селезенка, КМ,
ЦНС
Клетки Гоше
Болезнь Нимана – Сфингомиелиназа Печень,
Пика
селезенка, КМ,
ЦНС
Клетки Пика
Болезнь ТеяСакса
Гексозоминидаза
ЦНС, сетчатка
Изменение
глаза, селезенка, мейсснеровского
печень
сплетения в
кишке
35.
Болезнь Краббе36.
Болезнь Гоше37.
Мезенхимальные липидозы связаныс нарушением обмена нейтрального жира
и холестерина, приводят к развитию:
ожирения (общего и местного);
холестероза
(сосудов, желчного пузыря и др.).
38.
Виды ожирения1.
По этиологии:
2.
По распространенности:
3.
обменно-алиментарное;
эндокринное;
церебральное.
общее (симметричное);
по верхнему типу;
по нижнему типу.
По характеру (качеству):
гипертрофическое;
гиперпластическое;
смешанное.
39.
Ожирение и норма40.
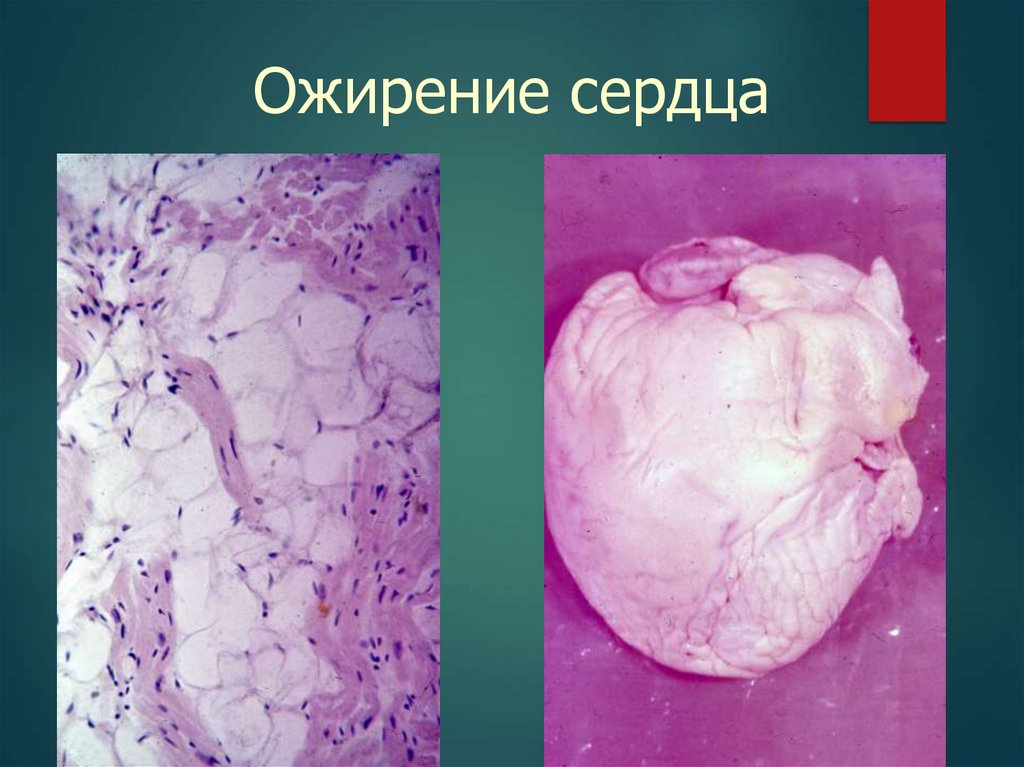
Ожирение41.
Ожирение сердца42.
Местный липоматоз► Болезнь
Деркума – образование в
подкожно-жировой
клетчатке узелков
из жировой ткани (болезненных при
пальпации).
► Болезнь
Маделунга – симметричное
образование жировых отложений на шее.
43.
Углеводные дистрофии1.
2.
3.
4.
Паренхиматозные (например, гликогенозы).
Мезенхимальные (например, нарушение обмена
муцина и мукоида – гликопротеинозы). Генетической
природы заболевание – муковисцидоз.
Приобретенные (например, сахарный диабет).
Наследственные (например, гликогеноз,
мукополисахаридоз, гликопротеиноз, муколипидоз).
44.
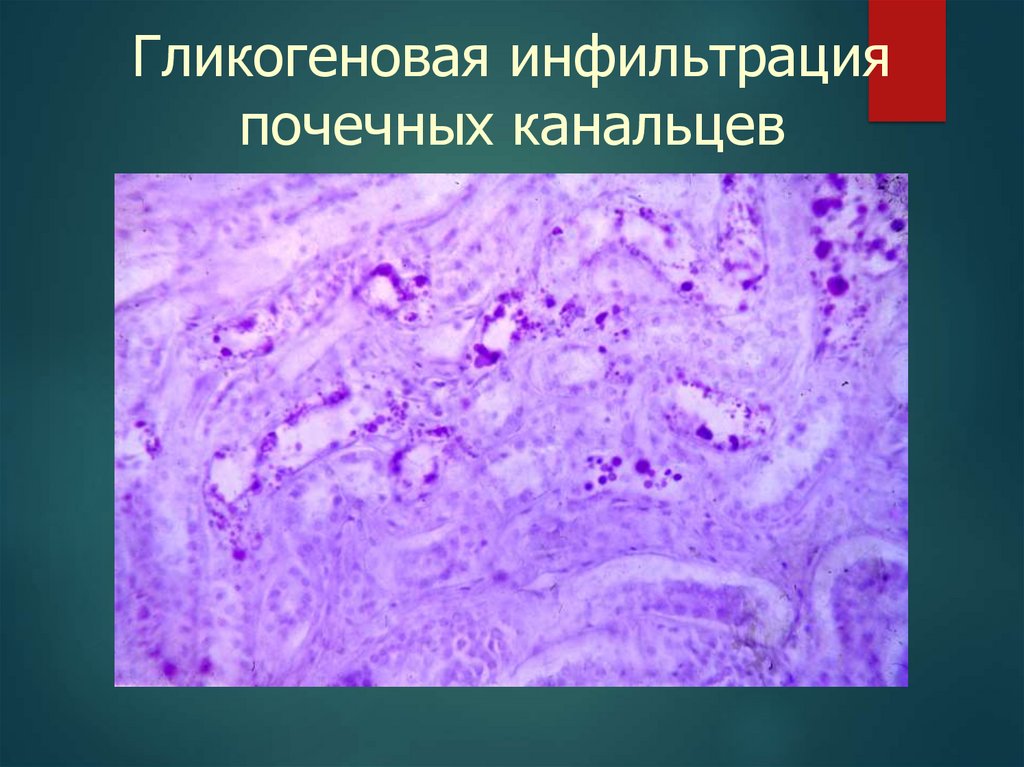
Гликогеновая инфильтрацияпочечных канальцев
45.
Муковисцидоз► Наследственное
системное заболевание
► Слизь густая и вязкая. Плохо выводится
из желез.
► Кистозное расширение желез с
образованием ретенционных кист и
фиброза
► Присоединение воспаления
46.
ГликогенозыНазвание
болезни
Дефицит
фермента
Локализация
накопления
гликогена
Гирке (1 тип)
Глукозо-6фосфатаза
Печень, почки
Помпе (2 тип)
Кислая αглюкозидаза
Мак-Ардля (5 тип) Фосфолипазы
И др.
миокард, мышцы
Скелетные мышцы
47.
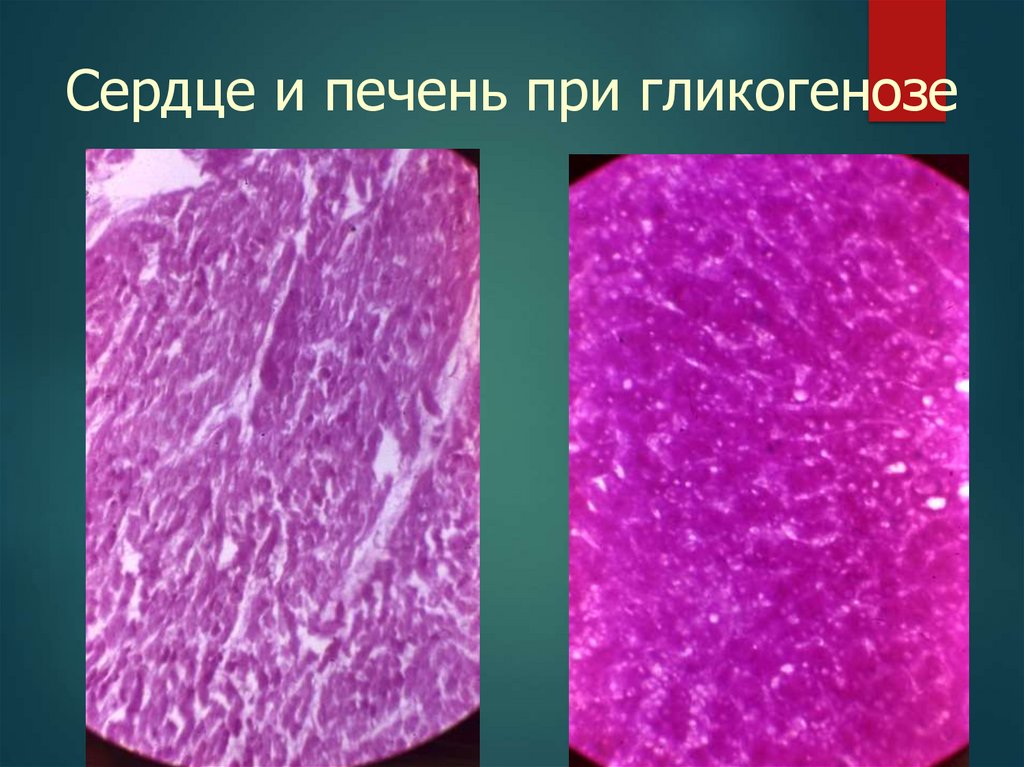
Сердце и печень при гликогенозе48.
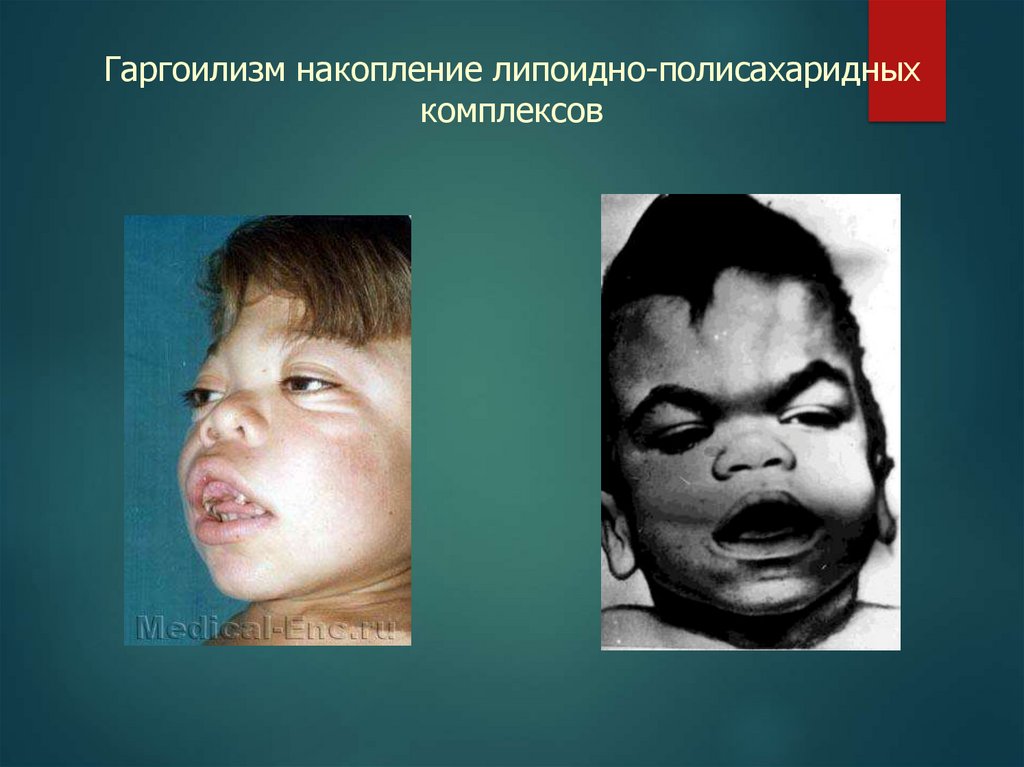
Гаргоилизм накопление липоидно-полисахаридныхкомплексов