










 из клетки- предшест-венницы миелопоэза, сохранившей способность")
 II А (эритремическая) – развернутая: миелоидная метаплазия селезенки, плеторический синдром, тромбозы. III")
 3. Дезагреганты (тиклид, пентоксифилин) 4. Цитостатики – в")

")



 Волосатоклеточный лейкоз")












 medicine
medicineSimilar presentations:






")



Дифференциальный диагноз лимфо- и миелопролиферативных заболеваний
1. ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ ЛИМФО- И МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ
Лектор – зав. каф. внутреннихболезней - проф. Л.М.Карзакова
2.
Миелопролиферативные заболевания:хронический миелолейкоз
эритремия
эритромиелоз
хр. мегакариоцитарный лейкоз (идиопатическая,
геморрагическая тромбоцитемия)
3.
Лимфопролиферативные заболевания →2 группы:хронические лимфоидные лейкозы и злокачественные лимфомы, имеющие первоначально
внекостномозговую локализацию (лимфатические
узлы, селезенка, кожа, лимфоидная ткань слизистой
желудка). При лейкозах опухолевый процесс первично
развивается в структурах КМ.
Лимфопролиферативные лейкозы:
хронический лимфолейкоз
Т-клеточные лейкозы
NK- клеточные лейкозы
4. Лейкозы – опухолевые клональные заболевания кроветворной системы с первичным поражением костного мозга
Лейкозы/
острые
↓
Потомки стволовой
кроветворной клетки
или клетки-предшественницы
\
хронические
↓
Зрелые или
созревающие
клетки
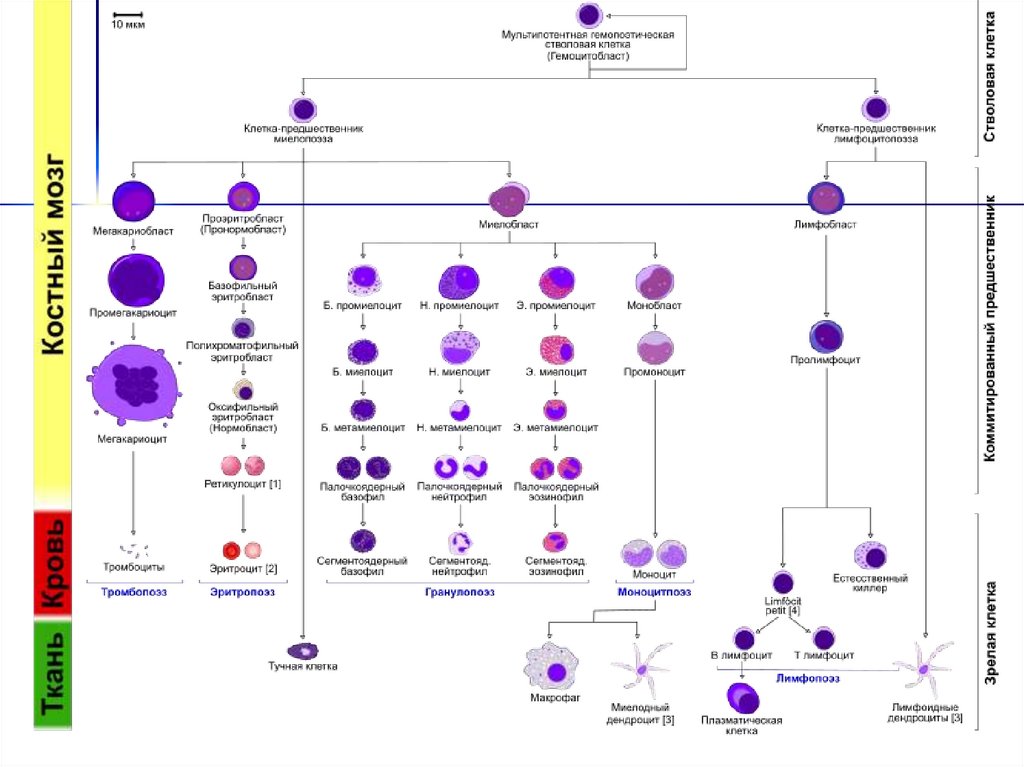
5. Схема кроветворения
Полипотентная стволовая клетка (СК)/
\
Лимфоидная СК
Миелоидная СК
/
\
/
|
\
Pro-B-л Кортик. Т-л
↓
↓
Pre-B-л Медуллярн.Т-л
↓
↓
Зрелые В-л Зрелые Т-л
↓
↓
Активир. Активир.
В-л
Т-л
CFU-G,M CFU-Meg
/
\
CFU-G CFU-M
BFU-E
↓
СFU-E
↓
Эритробласт
↓
Нормобласт
↓
Ретикулоцит
↓
Эритроцит
6. Этиопатогенез
Воздействие мутагенных факторов на геном:внешние (ионизирующая радиация, бензол, алкилирующие
цитостатики)
внутренние (повышенная чувствительность к внешним
онкогенным воздействиям)
Хромосомные аберрации (увеличение числа хромосом –
трисомия, делеция и/или транслокация участков
хромосом)
Экспрессия онкогенов или активация онкогенов →
стимуляторы пролиферации
Мутация генов апоптоза → угнетение апоптоза в
лейкозных клетках
7. Миелопролиферативные Хронический миелолейкоз
Из ранних клеток-предшественниц миелопоэза,сохраняющих способность дифференцироваться до
зрелых форм
субстрат – гранулоциты
2 стадии: развернутая-доброкачественная
(моноклоновая)
терминальная-злокачественная
(поликлоновая)
Заболеваемость – 1,0-1,5 на 100 000 человек в год,
средний возраст – 40-50 лет.
8.
9. Этиопатогенез
У 90-95 % больных хромосомная транслокация t (9 ↔22) собразованием Ph-хромосомы и в результате формируется
мутантный ген BCR-ABL, кодирующего белок р210
При переходе в терминальную стадию появляются клетки
с дополнительными аномалиями (вторая Ph-хромосомы)
Клиника
Развернутая ст.
Тяжесть и боль в левом подреберье (увеличение
селезенки)
Немотивированный нейтрофильный лейкоцитоз со сдвигом
до промиелоцитов, единичные бластные клетки, все
промежуточные формы гранулоцитов представлены,
базофильно-эозинофильная (↑) ассоциация. На ранних
стадиях – тромбоцитоз.
Астенический синдром, рост содержания мочевой кислоты
в крови и моче (из-за распада клеток)
10.
Терминальная ст.Повышается температура
Быстро растет селезенка
Сильные боли в костях
Саркомный рост в лимфоузлах
Опухолевая прогрессия (тромбоцитопения,
анемия, лимфоцитопения, гранулоцитопения,
бластные клетки↑ - бластный криз)
Лейкемиды в коже (из бластных клеток чаще)
Нейролейкемия - метастазированием лейкозных клеток в
оболочки головного и спинного мозга или в вещество мозга
Рефрактерность к цитостатикам
11. В крови бластный криз Костный мозг: бласты
12. ДИАГНОСТИКА
Трепанобиопсия с аспирацией костного мозга (увеличениечисла миелокариоцитов)
⇣ЩФ в гранулоцитах ПК
УЗИ: увеличение селезенки, печени, абдоминальных
лимфоузлов
Лечение
Гидроксимочевина 10-20 мг/кг
α – интерферон 5-9 млн Ед х 3 раза/нед в/м
Гливек (иматиниб мезилат) блокатор мутантного белка р210
400 мг/м2 - 28 дней – к гематологической ремиссии у 97%,
цитогенетической – у 82% - апоптоз клеток с Ph-хромосомой
Трансплантация КМ - моложе 50 лет в I стадии заболевания, в
70% случаев приводит к выздоровлению.
13. Эритремия истинная полицитемия (болезнь Вакеза) из клетки- предшест-венницы миелопоэза, сохранившей способность
Эритремияистинная полицитемия (болезнь Вакеза) из клетки- предшественницы миелопоэза, сохранившей способность дифференцироваться по 4 росткам, преимущественно по красному ростку
0,6-1,6 на 100 000 населения
Патогенез
Патология хромосом 20 пары, трисомия в 8-9 паре,
делеция 12 и 13 хромосом
Клиника
1.Начало постепенное: покраснение кожи, тяжесть в
голове, увеличение селезенки, артериальная
гипертония, мучительный зуд, эритромелалгии –
(из-за стазов в сосудах, повышенной агрегации
тромбоцитов → резкая боль,
красно-синюшный оттенок кожи).
2. Некрозы пальцев, тромбозы
сосудов, инсульт
14.
15. Стадии I (начальная) II А (эритремическая) – развернутая: миелоидная метаплазия селезенки, плеторический синдром, тромбозы. III
– анемическая – миелофиброз. Исход эритремии – острыйлейкоз
Диагностика
Подозрение: ♂ Эр > 5,7х1012/л, Нв >177 г/л, Ht > 52%;
♀ Эр> 5,2х1012/л, Нв >160 г/л, Ht > 50%.
Подтверждение: спленомегалия, лейкоцитоз > 12х109/л,
тромбоцитоз > 400х109/л, ↑ЩФ нейтрофилов.
Верификация: трепанобиопсия подвздошной кости (панмиелоз)
16. Лечение 1. Начинают с кровопусканий 2. Аспирин (40-80 мг в день) 3. Дезагреганты (тиклид, пентоксифилин) 4. Цитостатики – в
развернутой стадии при отсутствииэффекта от кровопусканий
гидроксимочевина 30 мг/кг в день внутрь – неделя,
затем поддерживающая доза 15 мг/кг.
5. α – интерферон 3-5 млн Ед х 3 раза/нед в/м
6. Симптоматическая терапия
Антигипертензионная
Свежезамороженная плазма – при острых тромбозах
сосудов
17.
Хронический мегакариоцитарный лейкозГипертромбоцитоз —более (1000—3000)х109/л. (иногда: 400600)х109/л. Для тромбоцитов характерны морфологические изменения
(гигантские, микро- и уродливые формы). Нет лейкоцитоза, сдвига
лейкоцитарной формулы влево, эритроцитоза либо они
выражены слабо.
Селезенка несколько увеличена; пальпируется у реберного
края.
Проявляется нарушениями гемостаза: 1) повышенной
наклонностью к тромбозам и в результате частыми сухими
некрозами концевых фаланг пальцев стоп, 2) наклонностью к
кровоточивости (чаще десен) и кровоподтекам (нарушение
агрегации тромбоцитов, повышенние местного фибринолиза,
ДВС-синдром с потреблением факторов гемостаза).
Эритромелалгии.
Лечение:
- дезагреганты (аспирин, курантил)
- цитостатик - гидроксимочевина (гидреа)
α-интерферон
Ингибитор фосфодиэстеразы III – анагрелид –
↓ образования Tr
Таргентная терапия - руксолитиниб → на
клетки с JAK2- мутацией
-
18.
19. Лимфопролиферативные Хронический лимфолейкоз (ХЛЛ)
90 % всех ХЛЛ – В-клеточный ХЛЛПроисходит из клетки-предшественницы В-клеток
Проявления – лейкоцитоз, лимфоцитарная
пролиферация в КМ, лимфоузлах, селезенке,
печени
Патогенез
Хромосомные аберрации (трисомия хромосом 12,
делеция 11 или 13). Патологические клетки
принадлежат малой субпопуляции
предшественников В-л (CD5+ ), разрастание
которых обусловливает основные признаки ХЛЛ.
Клиника
Многие годы – лишь лимфоцитоз. Постепенно
↑лимфоузлы, астенический синдром. Лимфоцитоз ↑до 8090%. На поздних стадиях – анемия, тромбоцитопения.
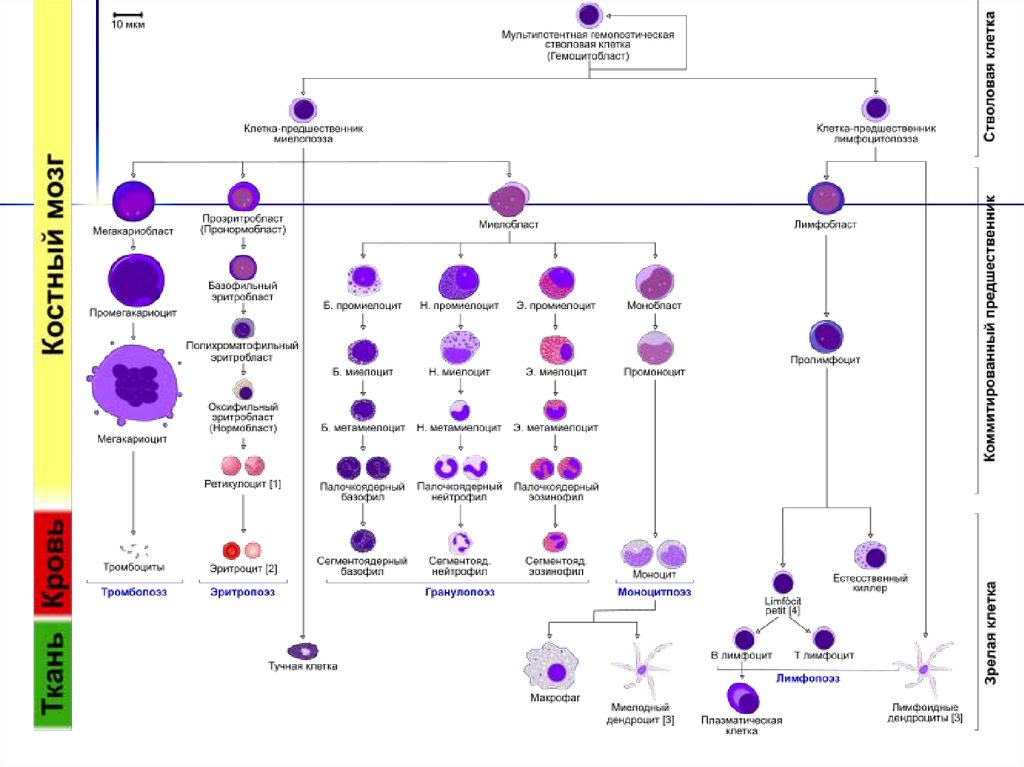
20. Схема кроветворения
Полипотентная стволовая клетка (СК)/
\
Лимфоидная СК
Миелоидная СК
/
\
/
|
\
Pro-B-л Кортик. Т-л
↓
↓
Pre-B-л Медуллярн.Т-л
↓
↓
Зрелые В-л Зрелые Т-л
↓
↓
Активир. Активир.
В-л
Т-л
CFU-G,M CFU-Meg
/
\
CFU-G CFU-M
BFU-E
↓
СFU-E
↓
Эритробласт
↓
Нормобласт
↓
Ретикулоцит
↓
Эритроцит
21. Диагностические критерии
Абсолютный лимфоцитоз в ПК (> 5х109/л)Морфология лимфоцитов:
- классический малый лимфоцит при
прогрессирующей форме
- лимфоцит с отростками – при волосатоклеточном
лимфолейкозе
Более 30 % лимфоцитов в пунктате КМ
Фенотип В-клеток CD19+, CD5+, CD23+.
22. Периферическая кровь : лимфоциты - 90 %, встречаются атипичные клетки с расщепленными ядрами, тени Боткина-Гумпрехта. Ядра
глыбчатые23. Периферическая кровь: лимфоциты - 94 % (из них 80 % - «волосатые» клетки) Волосатоклеточный лейкоз
24. Стадии
Начальная – лейкоциты 40-50х109/л, красныйросток, тромбоциты в норме, интоксикации
нет
Развернутая - лейкоциты > 40-50х109/л,
↓ массы тела, слабость, потливость,
цитолитические кризы (аутоиммунные
реакции – образование а/т к Er, Tr),
рецидивирующие инфекции
Терминальная – угнетение кроветворения,
кахексия, ↓Ig, инфекции (опоясывающий
лишай, туберкулез), нейролейкемия,
саркомный рост в л/у или в коже.
25. Лечение
3 программы ПХТФлударабин (антипуриновый перапарат) + митоксантрон
Флударабин + циклофосфан
Флударабин + циклофосфан + митоксантрон (наибольший
эффект)
Новое – мабтера (ритуксимаб) (МКАТ к CD20) – 375 мг/м2
еженедельно, на курс №8
Офатумумаб (Арзерра) – МКАТ к CD20
Трансплантация КМ
26. Т-клеточные опухоли
Из предшественников Т-лимфоцитовРедко встречаются
На опухолевых клетках CD1a, CD3
Принципы лечения
Как при остром лимфобластном лейкозе:
Винкристин
Преднизолон
Л-аспарагиназа
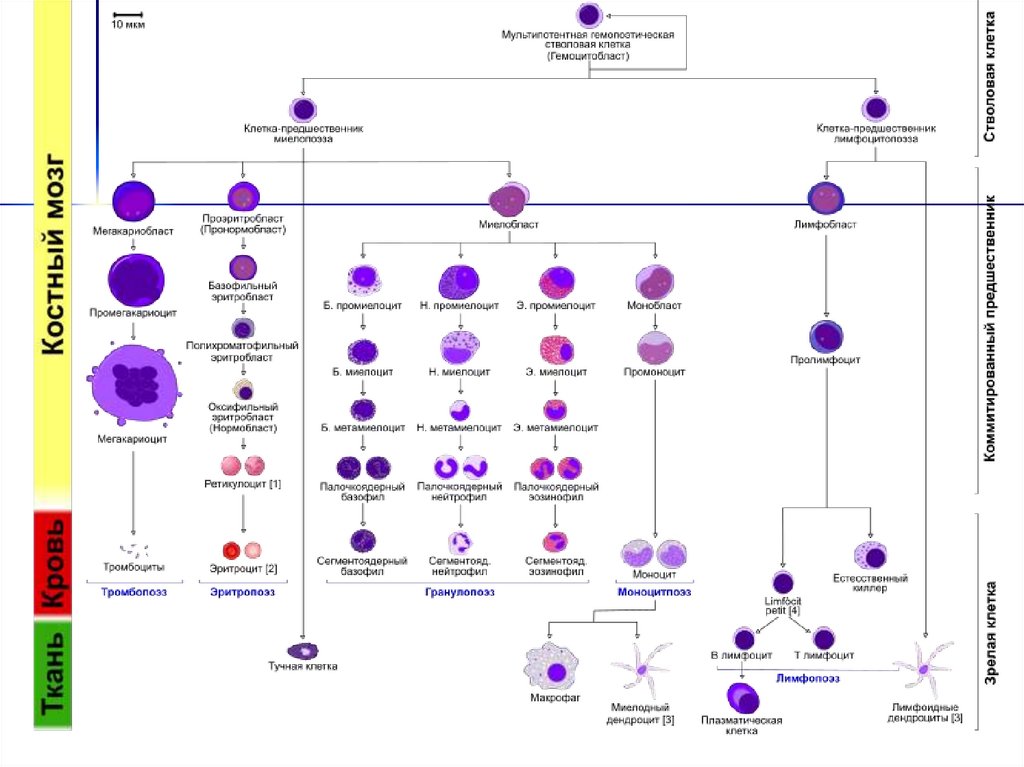
27. Схема кроветворения
Полипотентная стволовая клетка (СК)/
\
Лимфоидная СК
Миелоидная СК
/
\
/
|
\
Pro-B-л Кортик. Т-л
↓
↓
Pre-B-л Медуллярн.Т-л
↓
↓
Зрелые В-л Зрелые Т-л
↓
↓
Активир. Активир.
В-л
Т-л
CFU-G,M CFU-Meg
/
\
CFU-G CFU-M
BFU-E
↓
СFU-E
↓
Эритробласт
↓
Нормобласт
↓
Ретикулоцит
↓
Эритроцит
28.
ЛИМФОМЫ - опухоли лимфатических структур (лимфатические узлы,селезенка, вилочковая железа, кольцо Вальдейера, червеобразный
отросток, пейеровы бляшки)
Фолликулярная лимфома – В-клеточная лимфома с иммунофенотипом
CD20+, CD10+/-, BCL-2+, BCL-6+, CD3-, CD5-, CD23+/-, CD43-, cyclin D1-.
Лимфома маргинальной зоны – из клеток маргинальной зоны
лимфоидных фолликулов лимфатических узлов, селезенки и
лимфатических тканей (CD20+, CD5-, CD10-, CD23-, BCL-2+/-, BCL-6-,
MUM 1).
Лимфома из клеток мантии (CD20+, CD5+/CD43+, Cyclin D1+, BCL-2+,
CD3-, CD23) – агрессивная опухоль
Лимфома Беркитта – из зрелых В-лимфоцитов (CD20+, CD10+, CD38+,
BCL-6+, BCL-2-, CD44-, TdT-, CD3-) - агрессивная, на фоне
иммунодефицитов (трансплантация) - экстранодальная локализация
опухоли. Наиболее часто поражаются органы брюшной полости: тонкая
кишка (чаще ее терминальный отдел), брыжейка, а также желудок,
толстая кишка, брюшина, печень, селезенка.
29.
Т-клеточные лимфомы (CD4+/CD8-)30.
ЛИМФОГРАНУЛЕМАТОЗ (ЛИМФОМА ХОДЖКИНА)Субстрат – производные В-клеток зародышевых центров лимфатических
узлов - гигантские клетки Березовского–Штернберга–Рида (БШР), имеющие
2 и более ядра. Количество их в опухоли - 1–10 %. Клетки, имеющие схожую
с клетками БРШ морфологию, но одно ядро - клетки Ходжкина. (при
классическом варианте CD30+)
31.
КЛИНИКА• кашель, увеличение ЛУ, селезенки
• инфекционно-токсический синдром (лихорадка, ангины,
пневмонии, инфекции мочевых путей, вплоть до развития
септического состояния);
• геморрагический синдром (петехии, экхимозы, гематомы,
носовые и маточные кровотечения);
• исследование КМ: угнетение всех ростков кроветворения.
ЛЕЧЕНИЕ
• ХТ, лучевая терапия
32.
Иммуноглобулин-секретирующие лимфомы:• множественная миелома;
• солитарная плазмоцитома;
• макроглобулинемия Вальденстрема;
• лимфомы с моноклональной секрецией Ig;
• болезни тяжелых Ig;
• трудноклассифицируемые Ig-секретирующие опухоли.
33.
МНОЖЕСТВЕННАЯ МИЕЛОМА (плазмоцитома, болезньРустицкого-Калера) - В-клеточная злокачественная опухоль, субстрат плазматические клетки, продуцирующие моноклональный
иммуноглобулин. Согласно классификации ВОЗ 2017 г. термин
«множественная миелома» заменен на термин «плазмоклеточная
миелома» – заболевание, характеризующееся мультифокальной
пролиферацией неопластических плазматических клеток,
ассоциированное с секрецией моноклонального иммуноглобулина.
- Вялотекущая плазмоцитома (асимптоматическая)
- Критерии диагностики симптоматической ММ:
1.Плазматические клетки в КМ 10% и более
2. Моноклональный белок в сыворотке крови и/или в моче (за
исключением пациентов с несекретирующей ММ).
3. Один или более следующих признаков :
гиперкальциемия (2,65 ммоль/л),
почечная недостаточность (креатинин > 177 мкмоль/л),
анемия (Hb < 100 г/л),
поражение костей (остеолитические поражения, остеопороз или
патологические переломы).
34.
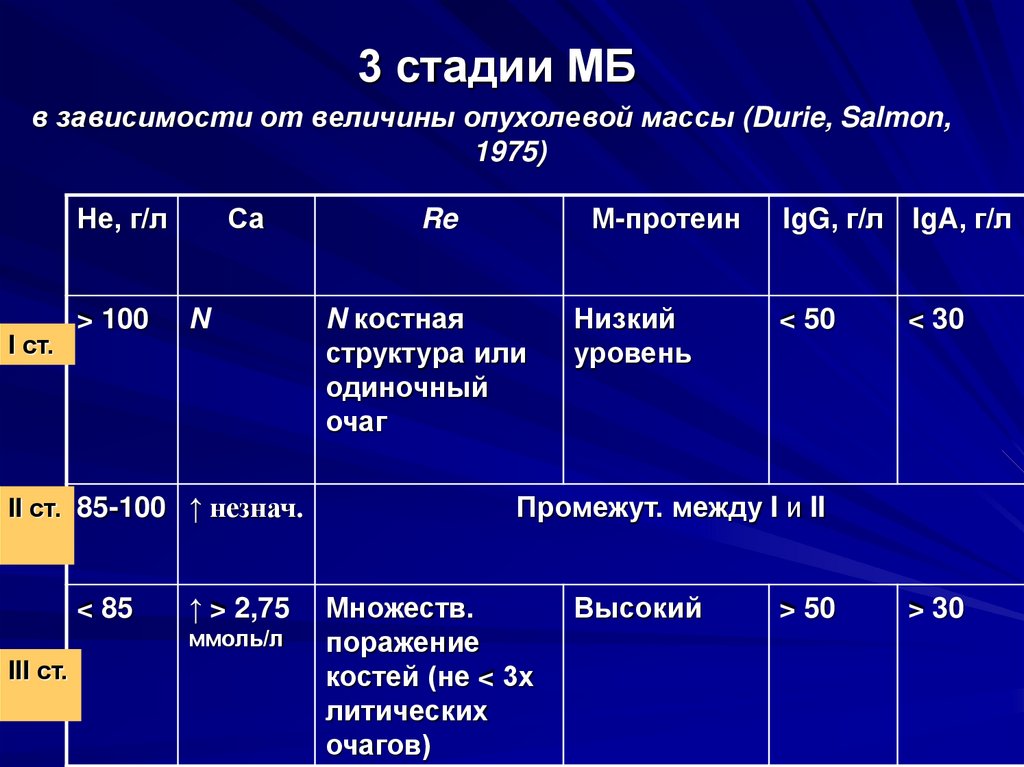
3 стадии МБв зависимости от величины опухолевой массы (Durie, Salmon,
1975)
Не, г/л
I ст.
> 100
Са
N
II ст. 85-100 ↑ незнач.
< 85
↑ > 2,75
ммоль/л
III ст.
Re
N костная
структура или
одиночный
очаг
М-протеин
IgG, г/л IgA, г/л
Низкий
уровень
< 50
< 30
Промежут. между I и II
Множеств.
поражение
костей (не < 3х
литических
очагов)
Высокий
> 50
> 30
35.
КЛИНИКА1. Пролиферация плазматических клеток
2. Секреция моноклональных антител
Синдром костной патологии
Поражение кроветворения
Гиперкальциемический синдром (тошнота,
рвота, слабость, запоры)
Синдром поражения почек (миеломная
нефропатия, амилоидоз, ХПН)
Синдром повышенной вязкости крови
Синдром белковой патологии
Неврологический синдром
Вторичный гуморальный ИД
36.
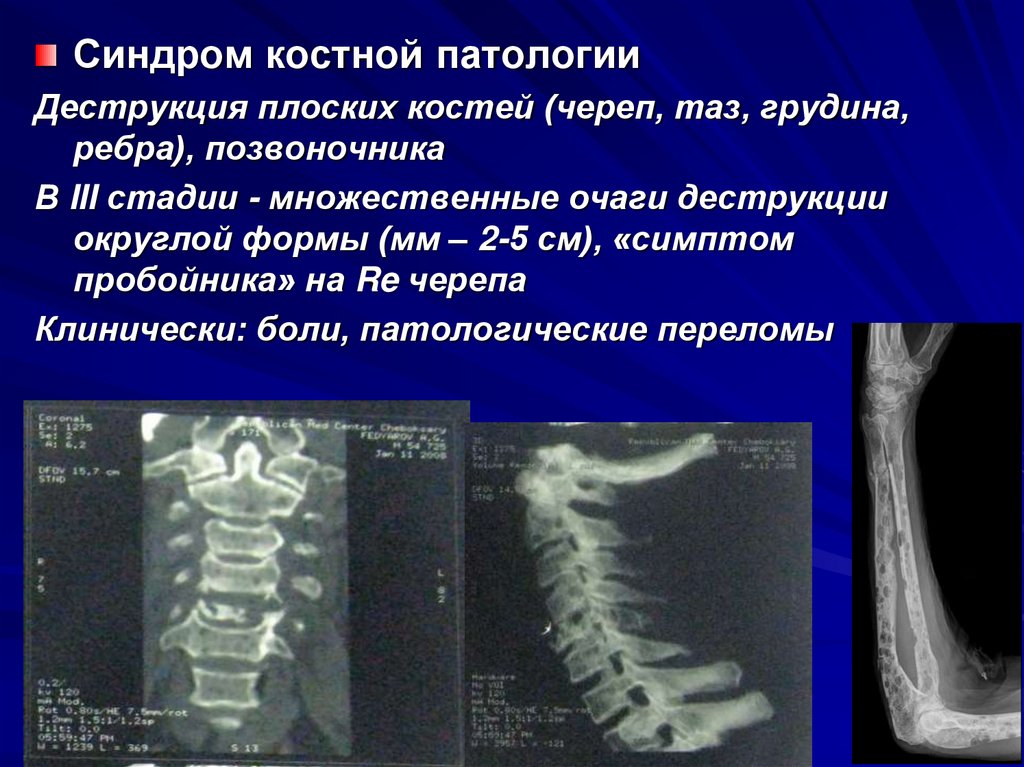
Синдром костной патологииДеструкция плоских костей (череп, таз, грудина,
ребра), позвоночника
В III стадии - множественные очаги деструкции
округлой формы (мм – 2-5 см), «симптом
пробойника» на Re черепа
Клинически: боли, патологические переломы
37.
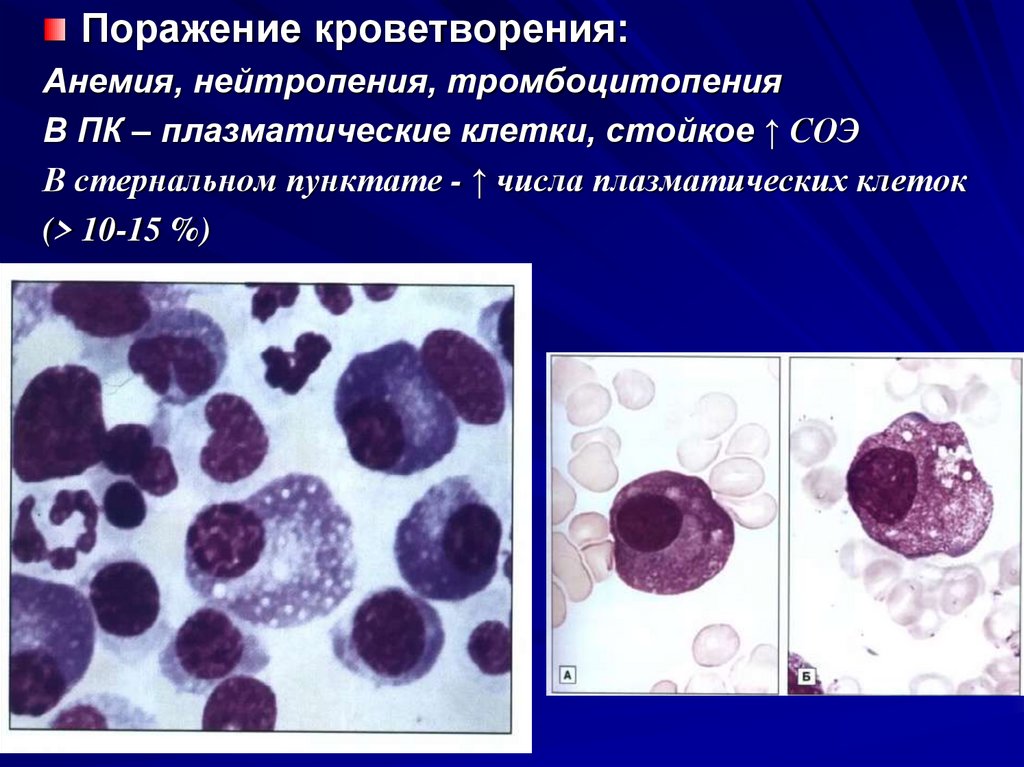
Поражение кроветворения:Анемия, нейтропения, тромбоцитопения
В ПК – плазматические клетки, стойкое ↑ СОЭ
В стернальном пунктате - ↑ числа плазматических клеток
(> 10-15 %)
38.
ЛЕЧЕНИЕ• основной метод – химиотерапия:
алкилирующие препараты: алкеран, мелфалан, сарколизин,
циклофосфан, препараты нитрозомочевины + их сочетание с
преднизолоном;
мелфалан + преднизолон (режим М + Р) – стандарт терапии
первой линии, 6-12 курсов → ремиссия → поддерживающая
терапия интерфероном-α в дозе 3 млн. ЕД/м2 3 раза в неделю,
но рецидивы неизбежны; можно использовать мелфалан в
высоких дозах;