






























 medicine
medicineSimilar presentations:










Болезнь Помпе и Нимана-Пика
1.
Болезнь Помпе и Нимана-ПикаВыполнила орд. Петрухина Е.А.
Кафедра Госпитальной педиатрии и
неонатологии СГМУ
2.
Болезнь Нимана-Пика - редкое наследственное нейровисцеральноезаболевание, вызываемое мутациями в генах SMPD1, NPC1 и NPC2 с
последующим нарушением внутриклеточного транспорта липидов и
накоплению холестерина и гликосфинголипидов в головном мозге и
других
тканях.
Развитие вариантов А и В болезни Ниманна-Пика (БНП-А, БНП-В)
связано с мутациями в гене сфингомиелинфосфодиэстеразы I (SMPDI), который кодирует фермент – кислую сфингомиелиназу (ASM).
Ген SMPD-I картирован на хромосоме 11, в локусе 11p15.4-p15.1. Развитие
типа
С
болезни
Ниманна-Пика
–
нарушение
структуры
трансмембранного белка, участвующего в переносе экзогенного
холестерина, которое связано с мутациями в гене NPC1 (локус 18q11-q12
хромосомы 18), ведущими к мутациям в гене NPC2 (локус 14q24
хромосомы
14)
и
приводящими
к
нарушению
структуры
холестеринсвязывающего белка.
3.
КлассификацияВ 1961 году, была предложена классификация заболевания:
- болезнь Ниманна-Пика, тип А: классическая инфантильная;
- болезнь Ниманна-Пика, тип В: висцеральная;
- болезнь Ниманна-Пика, тип С: неострая подростковая
- болезнь Ниманна-Пика, тип D: ново-шотландская;
Сегодня, когда понятна генетическая природа заболевания,
расстройство классифицируется:
- болезнь Ниманна-Пика, связанная с геном SMPD1, которая
включает в себя типы А и В;
- болезнь Ниманна-Пика, типа C, который включает в себя типы
C1 и C2. (Тип D возникает в результате мутации того же гена,
что и тип C1).
4.
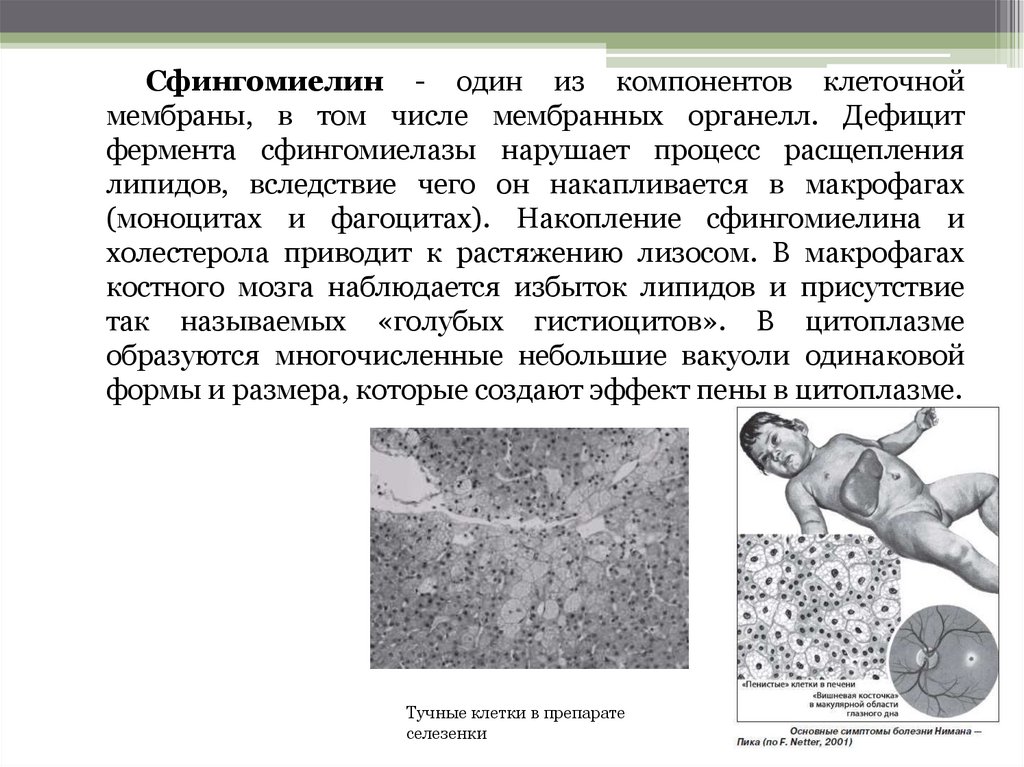
Сфингомиелин - один из компонентов клеточноймембраны, в том числе мембранных органелл. Дефицит
фермента сфингомиелазы нарушает процесс расщепления
липидов, вследствие чего он накапливается в макрофагах
(моноцитах и фагоцитах). Накопление сфингомиелина и
холестерола приводит к растяжению лизосом. В макрофагах
костного мозга наблюдается избыток липидов и присутствие
так называемых «голубых гистиоцитов». В цитоплазме
образуются многочисленные небольшие вакуоли одинаковой
формы и размера, которые создают эффект пены в цитоплазме.
Тучные клетки в препарате
селезенки
5.
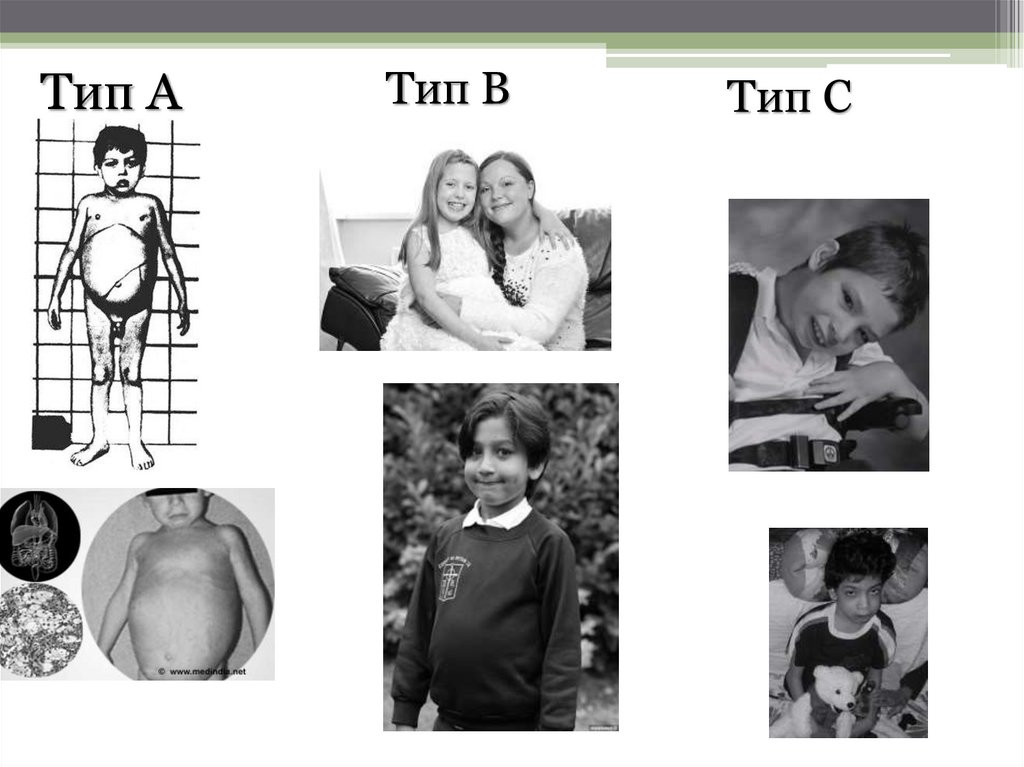
Тип АТип В
Тип С
6.
Тип А• чаще всего встречается у евреев, выходцев из Центральной и
Восточной части Европы
• липид сфингомиелин не расщепляется вовсе и быстро
наполняет клетки, вызывая нарушения их функций
• клетки увеличиваются в размере и быстро умирают, замещаясь
рубцовой тканью
• симптомы появляются в грудничковом возрасте – в 3-5
месяцев
• возникают трудности при вскармливании, они не набирают
вес, отстают в росте
• живот у таких деток пропорционально больше тела за счет
увеличения печени и селезенки
7.
Основные симптомы болезни Пика типа А:раннее начало развития болезни (3-5 месяцев),
рвота, частые поносы,
повышение температуры,
снижение массы тела, атрофия мышц, вялость,
постоянно увеличивается объем живота,
задержка психомоторного развития, (ребенок не перебирает
пальчиками, не садится),
• судороги,
• нарушается зрение, детки не фиксируют взгляд,
• снижается слух, дети плохо реагирую на звуки
Для типа А характерно быстрое прогрессирование симптомов
болезни. Пораженные клетки умирают, смерть клеток мозга
вызывает нарушения глотания, дыхания и кровообращения. Такие
дети редко доживают до 3-5 лет.
8.
Тип В• отсутствие накопления липидов в нервных клетках
• нервные клетки не разрушаются и не появляются симптомы
нарушения деятельности головного мозга
• не страдают интеллектуальные способности, в некоторых
случаях
больные
демонстрируют
довольно
высокую
умственную одаренность
Первые симптомы заболевания появляются после 3 лет. В
первую очередь у детей начинает увеличиваться селезенка и позже
печень. С возрастом появляются симптомы поражения легких.
Липид накапливается в лимфатических узлах и вызывает
снижение активности иммунной системы, дети часто болеют.
Длительность жизни таких больных несколько снижена, но они
доживают до взрослого возраста, иногда даже до старости.
9.
Проявления болезни Нимана-Пика типа В:• увеличение объема живота (за счет увеличения печени и
селезенки),
• периодические тупые боли в животе,
• тошнота, иногда рвота,
• нарушение работы печени и желчного пузыря (желтушность
кожи и глаз),
• повышенная кровоточивость (печень не вырабатывает
компоненты для свертывания крови в достаточном
количестве).
• одышка при умеренных физических нагрузках,
• частые респираторные инфекции и простуда.
10.
Тип С• проявляется после первых лет жизни
• в начале болезни происходит поражение внутренних
органов
–
увеличение
печени
и
селезенки,
лимфатических узлов
• поражаются и внутренние органы и нервная система.
Симптомы поражения внутренних органов:
• увеличение объема живота,
• ноющие, тупые боли в животе,
• желтушность кожи, слизистых оболочек и глаз,
• увеличение и болезненность лимфатических узлов,
• одышка,
• частые бронхиты и воспаления легких.
11.
• С течением болезни появляются симптомы поражения нервнойсистемы. Проявления нарушения роботы головного и спинного
мозга постоянно нарастают, больные отстают в психическом и
физическом развитии от сверстников. С прогрессированием
заболевания дети теряют навыки и умения, которыми уже
овладели. Например, ребенок уже научился разговаривать, но со
временем речь нарушается, становиться менее внятной.
• Поражение клеток нервной системы постоянно прогрессирует и
вызывает нарушения несовместимые с жизнью. Обычно такие
больные живут 15-20 лет.
12.
Симптомы поражения нервной системы:• тремор пальцев рук, нарушение координации
движений,
• судороги, эпилептические припадки,
• нарушения глотания и дыхания,
• потеря речи и других освоенных навыков,
• нарушения памяти и мышления, снижение
успеваемости в школе,
• нарушения поведения, замкнутость,
• эмоциональная нестабильность, раздражительность,
депрессия.
13.
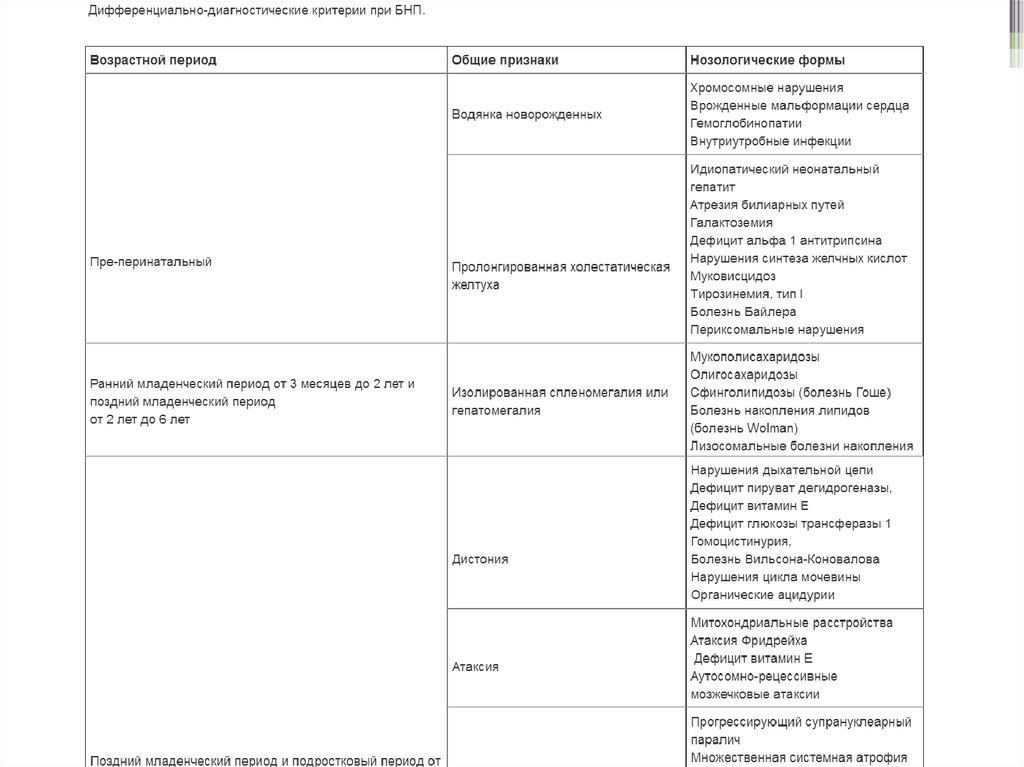
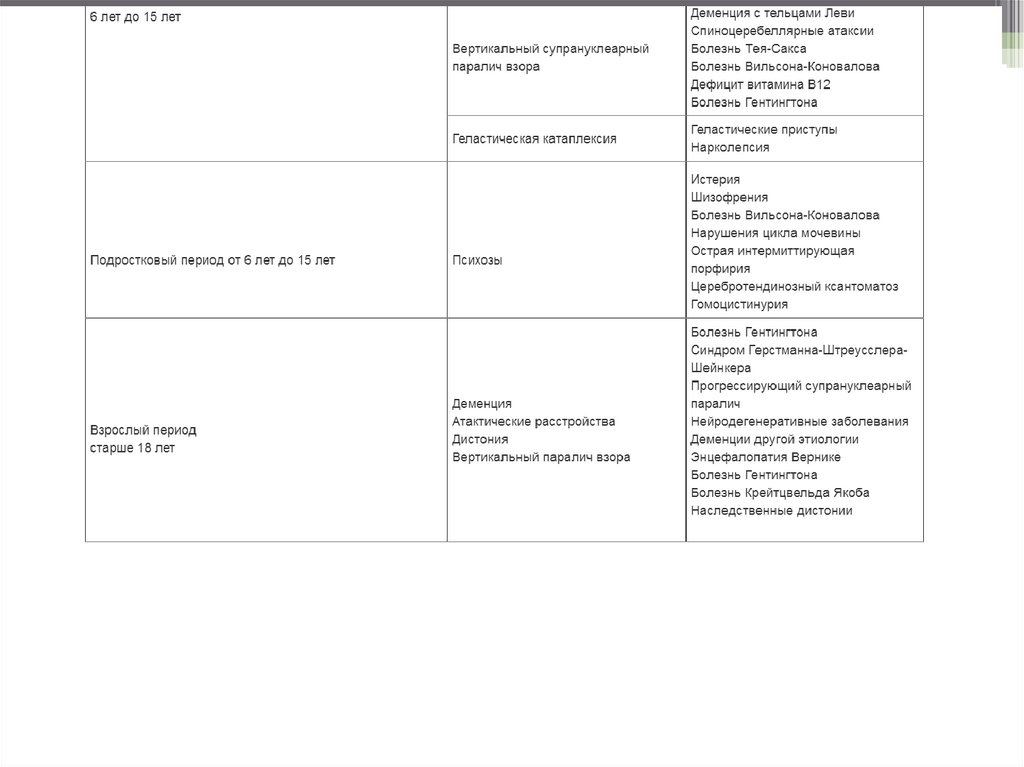
Основные (обязательные) диагностические обследования, проводимые на амбулаторном уровне:·
ОАК (развернутый);
·
ОАМ;
·
ЭКГ;
·
УЗИ печени;
·
УЗИ поджелудочной железы;
·
биохимический анализ крови (АЛТ, АСТ, билирубин, холестерин, ЛПНП, ЛПВП, триглицериды);
·
коагулограмма;
·
определение активности фермента хитотриозидаза в плазме крови.
Дополнительные диагностические обследования, проводимые на амбулаторном уровне:
·
рентгенограмма печени.
·
рентгенограмма легких.
Минимальный перечень обследования, который необходимо провести при направлении на плановую
госпитализацию: согласно внутреннему регламенту стационара с учетом действующего приказа уполномоченного органа в области
здравоохранения.
Основные (обязательные) диагностические обследования, проводимые на стационарном уровне
·
офтальмоскопия;
·
аудиометрия;
·
вызванные слуховые потенциалы;
·
ЭЭГ, видео ЭЭГ мониторинг (дневного/ночного сна);
·
МРТ головного мозга;
·
УЗИ органов брюшной полости (печени, поджелудочной железы);
·
биохимический анализ крови (АЛТ, АСТ, билирубин, холестерин, ЛПНП, ЛПВП, триглицериды);
·
определение активности фермента сфингомиелиназы методом масс-спектрометрии;
·
морфологическое исследование костного мозга (клетки Ниманна-Пика);
·
окраски филипином культуры фибробластов из биопсии кожи пациента, подтверждающий нарушения внутриклеточного
транспорта холестерина;
·
молекулярно-генетическое исследование (секвенирование) мутации в генах SMPDI, NPC1.
Дополнительные диагностические обследования, проводимые на стационарном уровне:
·
биопсия костного мозга;
·
бинокулярная электроокулография (регистрация саккадических движений глаз);
·
вызванные зрительные потенциалы
·
аудиография – определение остроты слуха;
·
ЭНМГ;
·
КТ головного мозга.
Диагностические мероприятия, проводимые на этапе скорой неотложной помощи: не проводятся.
14.
Лечение• снижение тяжести течения
• препараты, улучшающие функции печени и селезенки, отток
желчи
• препараты для улучшения работы нервной системы
Показано применение комплекса витаминов и минералов.
15.
Профилактические рекомендацииБольным обязательно назначают диету с ограничением употребления
определенных продуктов, таких как: черный хлеб, кукуруза, соки,
картофель.
Исключить:
• молочные продукты
• белый хлеб
• капуста
• рис
• сладости
• газированные напитки
• варенье
• бобовые
• огурцы
Без ограничений можно съедать гречку, все виды мяса, яйца, море
продукты, овощи и несладкие фрукты. Из сладких продуктов допускают
мед, травяные чаи, глюкозу, фруктозу.
Полностью излечить заболевание врачам пока не удается, но
правильно подобранная терапия может существенно снизить тяжесть
симптомов
и
улучшить
качество
жизни
больного.
16.
17.
18.
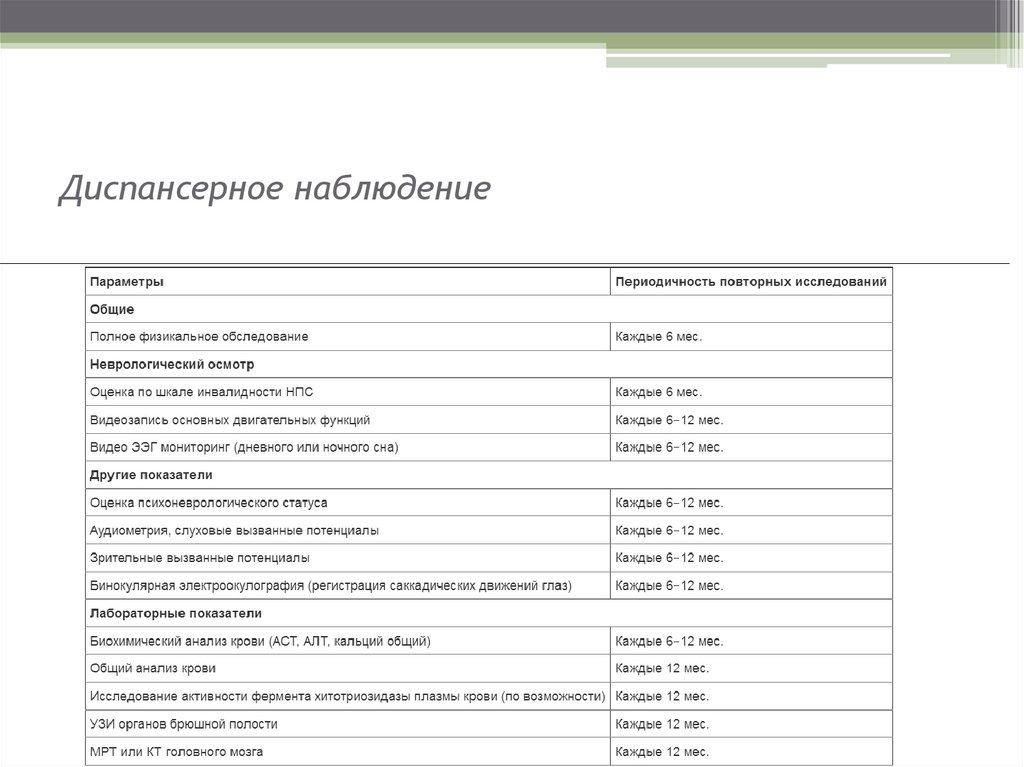
Диспансерное наблюдение19.
Клинический случайПациентка П., 11 лет, младшая дочь в неродственном браке здоровых родителей европейского происхождения. Сибс
(брат 17 лет) клинически здоров. Родилась от 3-й беременности, протекавшей с отслойкой плаценты в 12 нед,
хронической внутриутробной гипоксией плода. Роды на 41-й неделе, самопроизвольные, масса тела 3910 г, длина 51 см,
оценка по шкале АПГАР — 8/9 баллов. Раннее развитие: самостоятельно пошла в 1 год. Фразовая речь с 2 лет. С 3—4
лет появились неуклюжесть, тремор в руках. В 7 лет возникли трудности в обучении, атаксия конечностей. Была
проконсультирована в детском неврологическом центре с заключением: «синдромальная форма задержки развития,
тремор».
В 7 лет была проконсультирована в Институте коррекционной педагогики, диагноз: «выраженная задержка
психического развития; сенсомоторная алалия; дизартрия; экстрапирамидный синдром».
Кроме того, была направлена на консультацию врача-генетика с целью проведения дифференциальной диагностики с
наследственными атаксиями и дегенеративными заболеваниями ЦНС. Был выполнен хромосомный микроматричный
анализ, патогенного хромосомного дисбаланса выявлено не было, патогенные микроделеции и микродупликации
размером более 50 000 п.н. не были обнаружены. Участки потери гетерозиготности, содержащие гены, связанные с
феноменом импритинга, — отсутствовали. Протяженные участки потери гетерозиготности (>3 000 000 п.н.) — 0,11%
(общепопуляционный уровень).
В течение последующего времени наблюдалось прогрессирующее снижение интеллекта, нарастание нарушений
координации.
В 9 лет была проконсультирована клиническим психологом. Отмечены значительное снижение общего
интеллектуального развития, низкий запас знаний и представлений об окружающей среде, бедный словарный запас,
снижение объема внимания. В этом же возрасте развился первый приступ эпилепсии. Наблюдалась с диагнозом
криптогенная эпилепсия, проводимая терапия была неэффективна. В возрасте 11 лет появились признаки дисфагии,
патологическая поза левой ноги (ротация голеностопного сустава кнутри).
При консультации в Медико-генетическом научном центре в возрасте 11 лет была заподозрена БНП-С. При
неврологическом осмотре соматический статус был без существенных особенностей. Называла свое имя, возраст с
подсказкой. Периодически стереотипно подкашливала. Речь растянутая. Легкая гиперактивность. Поведение
корригировалось после нескольких повторений. Глазные щели и зрачки равной величины, фотореакции живые,
ограничение самопроизвольных движений глазных яблок вверх и при слежении за молоточком. Нистагма нет.
Асимметрии лицевой мускулатуры нет. Язык по средней линии. Мышечный тонус был умеренно снижен в
проксимальных отделах, дистальных отделах — дистоничный, в ногах — клонусы. Атетоидная установка кистей рук.
Гиперкинезы в руках. Сухожильная гиперрефлексия. Грубый интенционный тремор. Поперхивалась во время еды.
20.
• Биохимическое исследование плазмы и сухих пятен крови пациентки:концентрация триола 33,4 нг/мл (норма 2,0—50 нг/мг), концентрация 7кетохолестерина — 83,4 нг/мл (норма 10—75 нг/мг), активность хитотриозидазы —
51 нмоль/ч/мл (норма 2,5—100 нмоль/ч/мл).
• Молекулярно-генетическое исследование: проведен полный анализ гена NPC1,
методом
прямого
автоматического
секвенирования
проанализированы
кодирующие экзоны гена 1—25 и прилегающие интронные участки. Выявлена
описанная в базах данных мутация с.3070C>T (p.Pro1007Ala) в 20 экзоне гена в
гетерозиготном состоянии и неописанная замена с.3148G>А (p.Asp1050Asn) в 21
экзоне гена в гетерозиготном состоянии. Проведен анализ патогенности замены
c.3148G>A: базы MutationTaster, PolyPhen-2, PROVEAN, SIFT — патогенная замена.
Проведено молекулярно-генетическое исследование крови родителей и брата
пробанда: у матери мутация с.3148G>А в гетерозиготном состоянии (носитель), у
отца — с.3070С>G в гетерозиготном состоянии (носитель), у брата — с.3070С>G в
гетерозиготном состоянии (носитель).
21.
Ночной видео-ЭЭГ-мониторинг: основная активность сформирована в пределах возрастной
нормы, дезорганизована низкочастотными волнами. В бодрствовании частотные
характеристики основной активности в затылочных отделах полушарий головного мозга в
пределах возрастной нормы — 8—8,5 Гц. В бодрствовании и во сне регистрируется преходящее
региональное тета-, дельта-замедление в правой височно-теменно-затылочной области, реже в
левой лобно-центрально-височной области. Устойчиво регистрируются мультирегиональные
эпилептиформные изменения в виде комплексов пик-медленная волна, острая-медленная
волна в левой затылочной области с тенденцией к латерализации на все левое полушарие; в
правой лобно-центрально-височной области, независимо в левой задне-лобно-центральной
области с распространением на задние отделы соименного полушария и дистантным
распространением на гомологичные отделы противоположного полушария, а также короткие
диффузные низкосинхронизированные разряды комплексов острая—медленная волна чаще с
акцентом в правом полушарии, длительностью не более 0,1—0,5 с. Представленность
диффузных эпилептиформных разрядов уменьшается во сне. В целом в бодрствовании и во сне
индекс представленности эпилептиформной активности достигает 35—40% на продолженных
эпохах записи.
Ультразвуковое исследование внутренних органов — умеренно выраженная спленомегалия.
На МРТ головного мозга, выполненной в возрасте 11 лет, выявляется диффузная церебральная
атрофия, задержка миелинизации перивентрикулярного белого вещества задних отделов
больших полушарий, отмечается отрицательная динамика по сравнению с предыдущими
исследованиями.
Клинический диагноз: БНП-С (гликосфинголипидоз, лизосомная болезнь накопления
липидов), поздняя младенческая форма, аутосомно-рецессивный тип наследования.
Симптоматическая мультифокальная эпилепсия с ороалиментарными автоматизмами,
вторично-генерализованными судорожными приступами, фокальными диалептическими
приступами.
22.
• В данном наблюдении имело место постепенное начало заболевания при отсутствиигрубых
изменений
головного
мозга,
неэффективность
проводимой
противоэпилептической
терапии,
выявленная
«деликатная»
спленомегалия,
вертикальный супрануклеарный паралич взора. Сочетание задержки психомоторного
развития, полиморфной неврологической симптоматики и соматических изменений в
виде изолированной спленомегалии позволили предположить у пациентки БНП-С.
• Сложность диагностики заболевания в более раннем возрасте заключается в трудностях
объединения неспецифических клинических проявлений БНП-С, таких как задержка
развития, эпилепсия, неуклюжесть, в единый симптомокомплекс одного заболевания.
• В помощь практическим врачам для облегчения клинической диагностики БНП-С F.
Wijburg и соавт. предложили балльный диагностический индекс вероятности БНП-С.
Каждому симптому заболевания присваивается определенное количество баллов в
зависимости от уровня риска. При общей сумме баллов <40 — диагноз БНП-С
маловероятен, 40—69 — необходимо дополнительное обследование в центре БНП-С,
≥70 — необходимо направить пациента для проведения лабораторной диагностики на
БНП-С. У данной пациентки индекс вероятности составил 88 баллов.
• В настоящее время разработана патогенетическая субстрат-редуцирующая терапия для
БНП-С. Единственным разрешенным в ряде стран лекарственным препаратом для
лечения БНП-С является миглустат. Механизм действия субстрат-редуцирующей
терапии — ингибирование фермента глюкозилцерамидсинтазы, отвечающего за
первый этап синтеза большинства гликосфинголипидов (уменьшает их накопление в
клетке). Своевременная диагностика заболевания и назначение специфической и
симптоматической терапии увеличивают продолжительность жизни больных и
улучшают ее качество. Пациенты должны получать субстрат-редуцирующую терапию
пожизненно. Больной П., представленной в клиническом наблюдении, был назначен
специализированный препарат сразу после подтверждения диагноза.
23.
• Болезнь Помпе (БП), также широко известная как гликогеноз IIтипа, относится к редким мультисистемным наследственным болезням
накопления, связанным с дефицитом фермента кислой мальтазы
(кислой альфа-глюкозидазы) в лизосомах. Преимущественное
накопление гликогена отмечено в скелетных мышцах, но в разной
степени может обнаруживаться и в других органах и тканях, включая
сердечную мышцу, печень, нервную систему, гладкую мускулатуру и
т.п.
Характерен выраженный клинический полиморфизм
Выделяют две клинические формы:
1. Младенческая форма ( частота 1:38 000 новорожденных)
2. Поздняя форма ( частота 1:57000 новорожденных)
* Дополнительно описана «кардиальная форма», в тех случаях, когда
ведущими клиническими симптомами является нарушение со стороны
сердца.
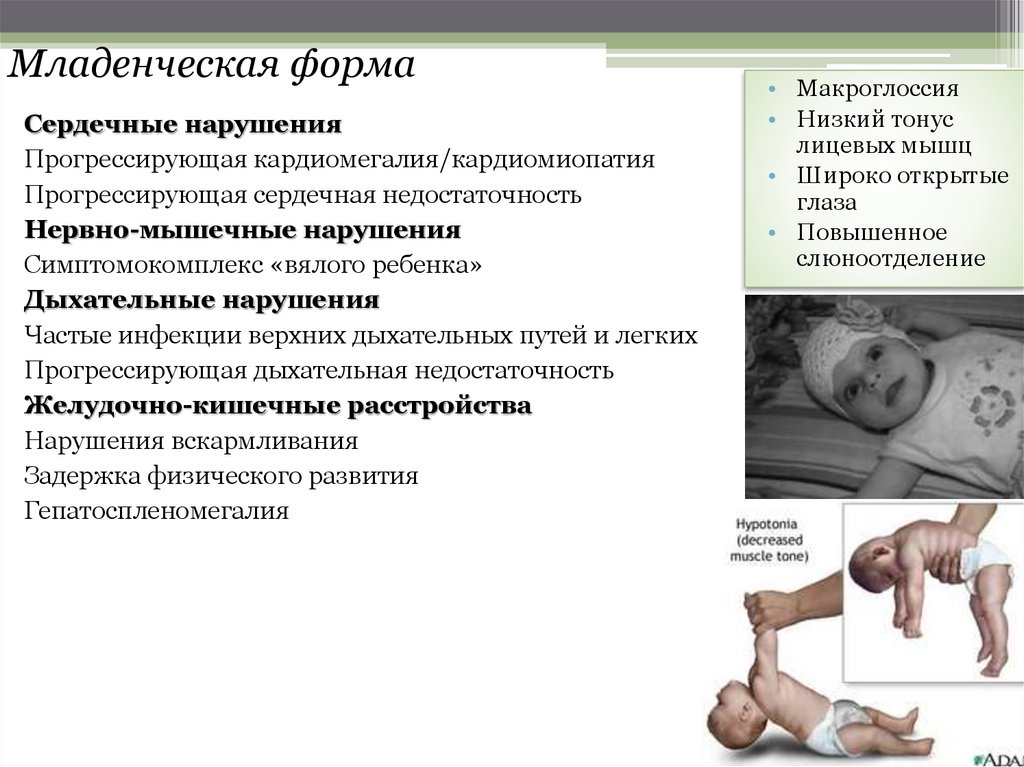
24.
Младенческая формаСердечные нарушения
Прогрессирующая кардиомегалия/кардиомиопатия
Прогрессирующая сердечная недостаточность
Нервно-мышечные нарушения
Симптомокомплекс «вялого ребенка»
Дыхательные нарушения
Частые инфекции верхних дыхательных путей и легких
Прогрессирующая дыхательная недостаточность
Желудочно-кишечные расстройства
Нарушения вскармливания
Задержка физического развития
Гепатоспленомегалия
• Макроглоссия
• Низкий тонус
лицевых мышц
• Широко открытые
глаза
• Повышенное
слюноотделение
25.
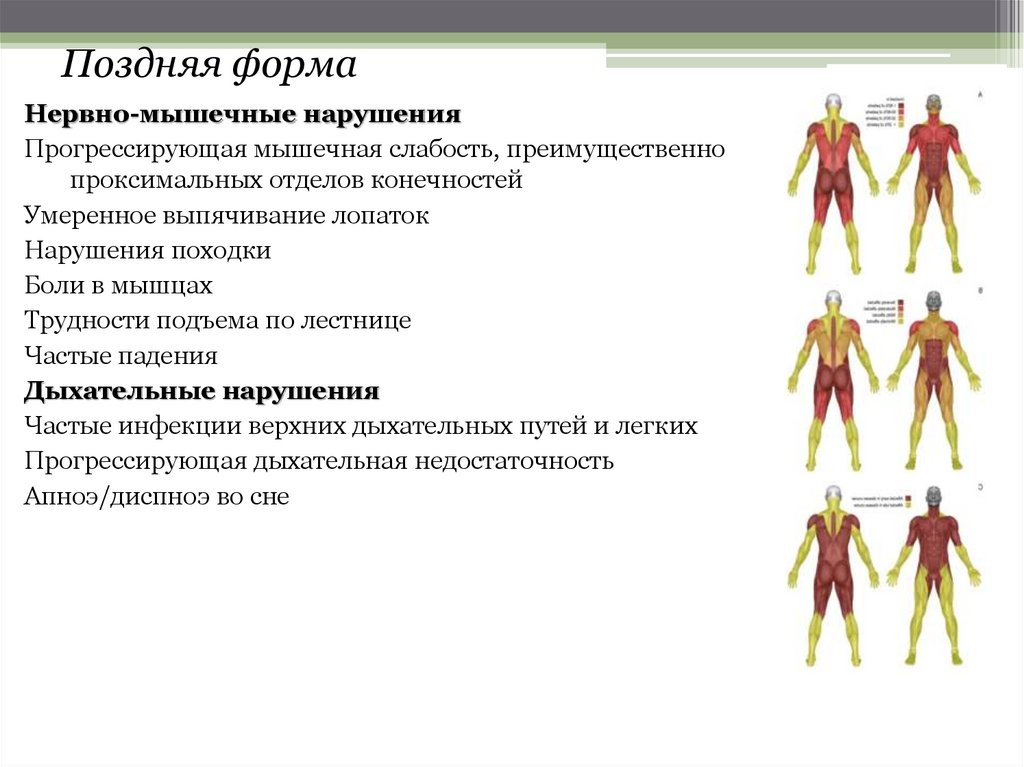
Поздняя формаНервно-мышечные нарушения
Прогрессирующая мышечная слабость, преимущественно
проксимальных отделов конечностей
Умеренное выпячивание лопаток
Нарушения походки
Боли в мышцах
Трудности подъема по лестнице
Частые падения
Дыхательные нарушения
Частые инфекции верхних дыхательных путей и легких
Прогрессирующая дыхательная недостаточность
Апноэ/диспноэ во сне
26.
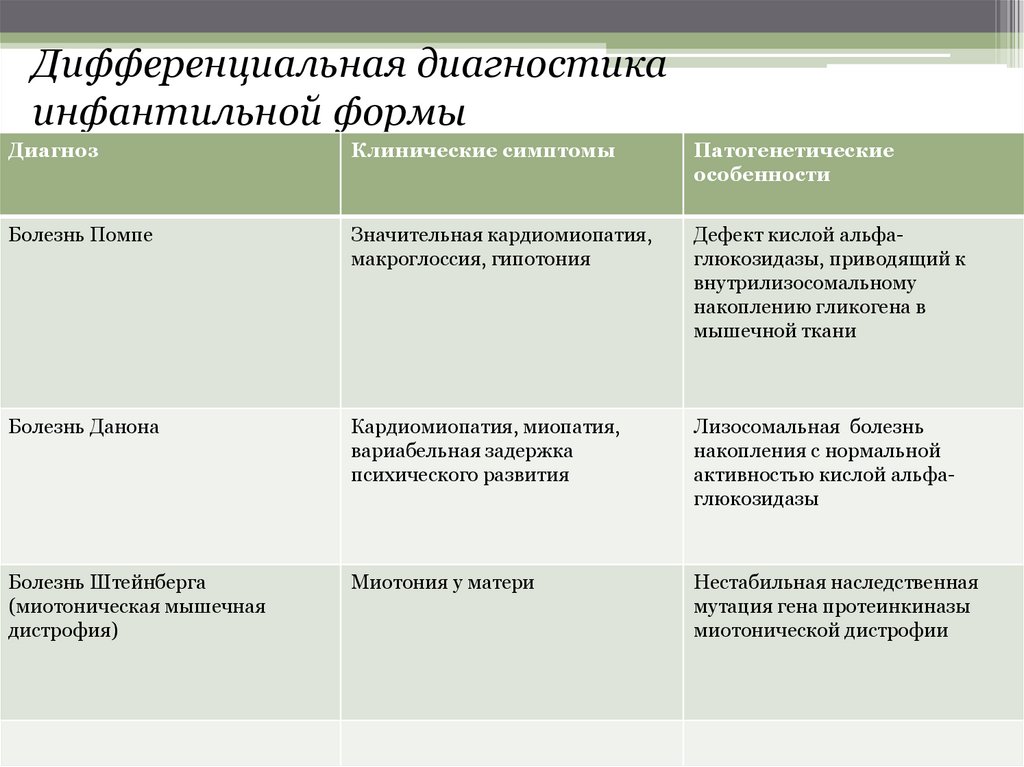
Дифференциальная диагностикаинфантильной формы
Диагноз
Клинические симптомы
Патогенетические
особенности
Болезнь Помпе
Значительная кардиомиопатия,
макроглоссия, гипотония
Дефект кислой альфаглюкозидазы, приводящий к
внутрилизосомальному
накоплению гликогена в
мышечной ткани
Болезнь Данона
Кардиомиопатия, миопатия,
вариабельная задержка
психического развития
Лизосомальная болезнь
накопления с нормальной
активностью кислой альфаглюкозидазы
Болезнь Штейнберга
(миотоническая мышечная
дистрофия)
Миотония у матери
Нестабильная наследственная
мутация гена протеинкиназы
миотонической дистрофии
27.
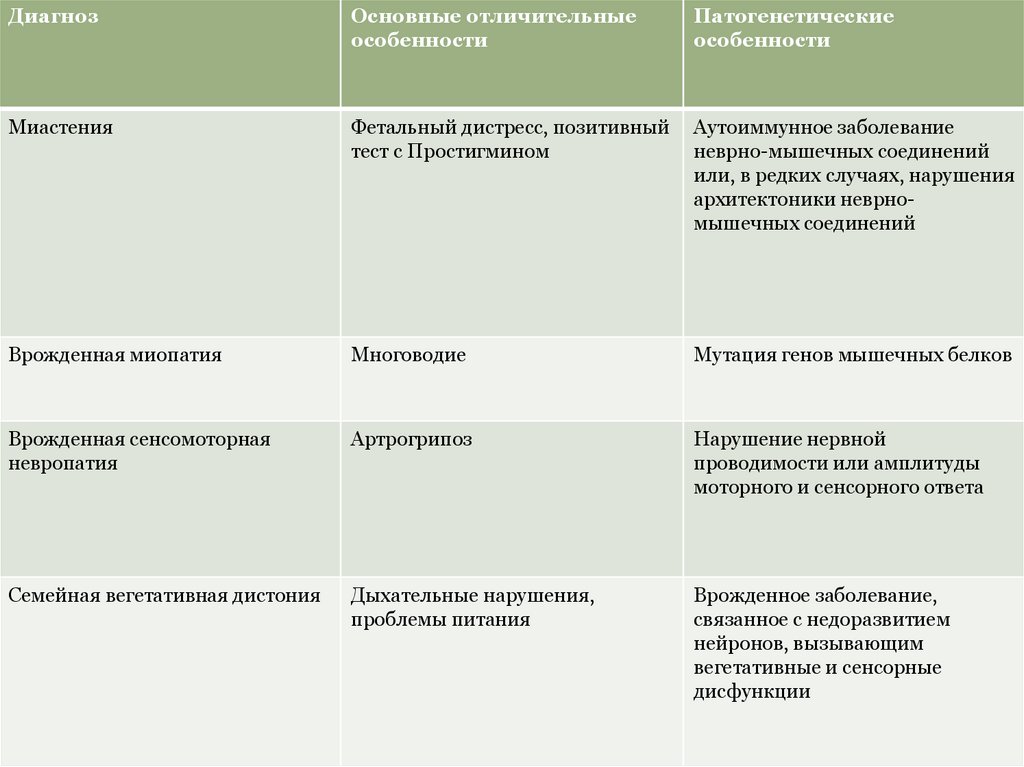
ДиагнозОсновные отличительные
особенности
Патогенетические
особенности
Миастения
Фетальный дистресс, позитивный
тест с Простигмином
Аутоиммунное заболевание
неврно-мышечных соединений
или, в редких случаях, нарушения
архитектоники неврномышечных соединений
Врожденная миопатия
Многоводие
Мутация генов мышечных белков
Врожденная сенсомоторная
невропатия
Артрогрипоз
Нарушение нервной
проводимости или амплитуды
моторного и сенсорного ответа
Семейная вегетативная дистония
Дыхательные нарушения,
проблемы питания
Врожденное заболевание,
связанное с недоразвитием
нейронов, вызывающим
вегетативные и сенсорные
дисфункции
28.
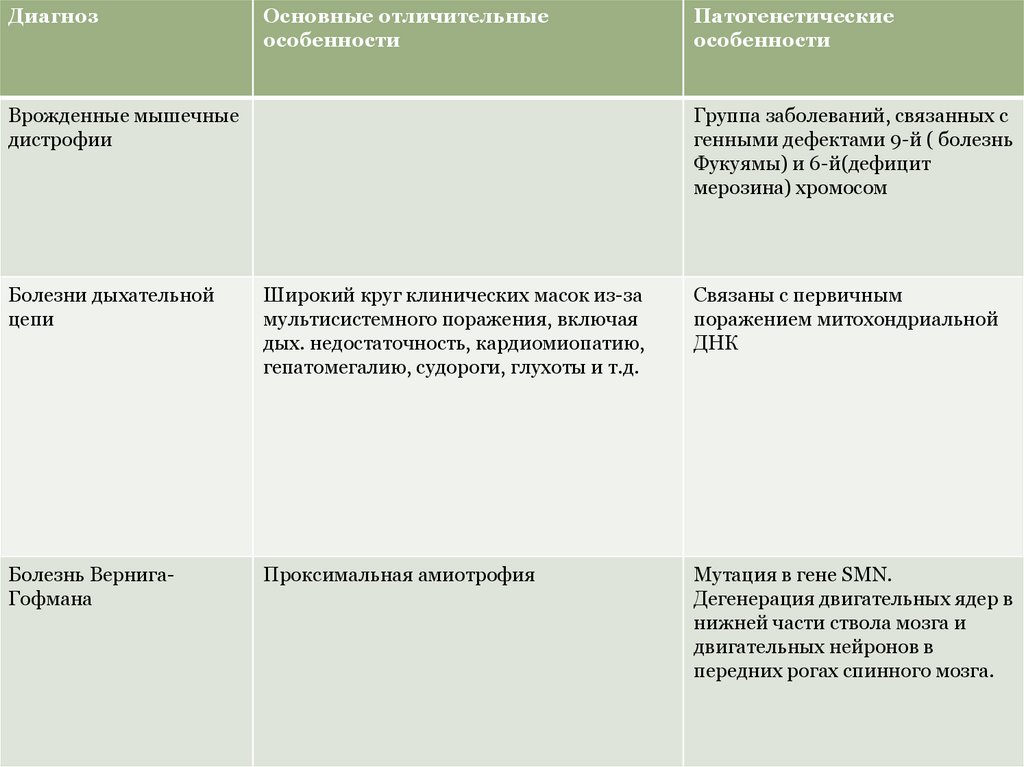
ДиагнозОсновные отличительные
особенности
Врожденные мышечные
дистрофии
Патогенетические
особенности
Группа заболеваний, связанных с
генными дефектами 9-й ( болезнь
Фукуямы) и 6-й(дефицит
мерозина) хромосом
Болезни дыхательной
цепи
Широкий круг клинических масок из-за
мультисистемного поражения, включая
дых. недостаточность, кардиомиопатию,
гепатомегалию, судороги, глухоты и т.д.
Связаны с первичным
поражением митохондриальной
ДНК
Болезнь ВернигаГофмана
Проксимальная амиотрофия
Мутация в гене SMN.
Дегенерация двигательных ядер в
нижней части ствола мозга и
двигательных нейронов в
передних рогах спинного мозга.
29.
Дифференциальная диагностикапозднодебютирующих форм
Тип заболевания
Диагнозы
Миодистрофии
Конечностно-поясные миодистрофии. Дистрофинопатии (Дюшена и
Беккера). Миофибриллярные миопатии. Миотоническая дистрофия 2-го
типа. Лопаточно-перонеальные синдромы. Болезнь Данона.Хсфепленная миопатия с повышенной аутофагией. Плечелопаточнолицевая миодистрофия
Воспалительные миопатии
Центрально-стержневая и многостержневая миопатии. Центральноядерная миопатия. Миопатия с гиалиновыми тельцами.
Другие метаболические
миопатии
Дефицит фермента, предотвращающего ветвление гликогена (
гликогеноз III). Дефицит ветвящего фермента гликогена ( гликогеноз IV).
Болезнь Мак Ардла ( с поздним дебютом). Митохондриальная миопатия.
Миопатия с наршуениями липидного обмена.
30.
Тип заболеванияДиагнозы
Болезни моторных нейронов
Спинальная мышечная атрофия, типы I и III.
Болезнь Кеннеди. Боковой амиотрофический
склероз.
Болезни нервно-мышечных соединений
Миастения. Врожденные миастенические
синдромы. Синдром Ламберта-Итона
31.
Лабораторные методы диагностики• Определение «косвенных» маркеров болезни Помпе
• Определение активности фермент альфаглюкозидазы
• ДНК-диагностика
32.
Косвенные маркеры болезни ПомпеПовышение активности КФК
• Наблюдается у большинства пациентов
• Редко превышает 2000 Ед/Мл
• Определяется практически во всех лабораториях
Повышение активности АЛТ, АСТ, ЛДГ
• Крайне неспецифичные макреры
• Определяется во всех лабораториях
Повышение глюкозо-тетрасахарида в моче
33.
Ферментный тест с использованием сухогопятна крови:
Ген GAA
• Описано более 200 различных мутаций.
• Мутация IVSI-13T-G – одна из наиболее частых при
взрослой и юношеской форме болезни
34.
Лечение болезни Помпе• Ферментзаместительная терапия (препарат
Myozyme). Показано внутривенное введение
препарата в дозе 20 мг/кг каждые две
недели
• Симптоматическое лечение
Прогноз болезни зависит от возраста дебюта заболевания, количества
вовлеченных систем организма, включая степень и тяжесть поражения
мышечного аппарата (скелетной, дыхательной, сердечной мускулатур), скорости
прогрессирования, времени начала ФЗТ. Назначение патогенетической терапии
на ранних стадиях заболевания определяет благоприятный прогноз и улучшает
качество жизни пациентов с болезнью Помпе, предотвращая их инвалидизацию.
Профилактики болезни Помпе не существует.
Профилактика осложнений:
• Рекомендована вакцинация по индивидуальному графику, которая включает в
себя вакцины, включенные в национальный календарь прививок, а также вакцину
против респираторно-синцитиального вируса человека, сезонную вакцину от
гриппа, пневмококковую вакцину. Диагноз МБП не является противопоказанием
для проведения прививок
35.
Клинический случайМиша С., 5мес. Из анамнеза известно, что ребенок родился от первой
беременности, протекавшей с анемией и нефропатией в I и II триместрах. У отца
пробанда — гипертрофия миокарда левого желудочка (ЛЖ). Роды срочные,
быстрые, осложненные тугим однократным обвитием пуповины вокруг шеи.
Безводный промежуток — 5 ч. Масса тела при рождении 3480 г, длина 54 см,
окружность головы (о.г.) 36 см, окружность груди 36 см. Оценка по шкале Апгар
7/8 баллов. Закричал после отсасывания слизи. Состояние при рождении средней
тяжести за счет неврологической симптоматики в виде синдрома угнетения
центральной нервной системы с элементами возбуждения и общего отечного
синдрома. В возрасте 2 нед поступил в отделение патологии новорожденных
Перинатального кардиологического центра ГКБ № 67 в связи с брадикардией с
частотой сердечных сокращений (ЧСС) до 80 в 1 мин.
36.
Состояние ребенка при поступлении было тяжелым за счет неврологическойсимптоматики в виде полного отсутствия двигательной активности, диффузной мышечной
гипотонии, спонтанного клонуса стоп, тремора ручек, оперкулярных пароксизмов,
гипорефлексии. Умеренно диспластичный фенотип: череп вытянут в передне-заднем
направлении, неправильная форма ушных раковин. Дыхание поверхностное, макроглоссия.
Со стороны сердечно-сосудистой системы отмечалось расширение границ относительной
сердечной тупости в обе стороны, приглушение тонов сердца, брадиаритмия. Печень: +2,0
см. В результате проведенного обследования у ребенка была выявлена гипертрофия
миокарда ЛЖ без обструкции выводного отдела ЛЖ. Показатели нейросонографии
демонстрировали наличие выраженной дилатации передних рогов боковых желудочков
мозга (Б=12 мм, ^ = 10 мм) и затылочных рогов (В=Б=25мм) с быстрым нарастанием
размеров окружности головы и отрицательной динамикой показателей нейросонографии.
Имел место эпизод фебрильных судорог.
С учетом мышечной гипотонии, брадикардии, макро-глоссии, субиктеричного оттенка
кожных покровов проведена оценка тиреоидного статуса (тиреотропный гормон, Т3, Т4 в
сыворотке крови) и исключен врожденный гипотиреоз. В возрасте 2 мес жизни сохранялся
гидроцефальный синдром (о. г. 39,5 см), диффузная мышечная гипотония (голову не
держит) и макроглоссия. С этого же возраста ребенок начинает часто болеть (острые
респираторные вирусные инфекции, бронхиты с бронхообструктивным синдромом,
бронхиолиты, пневмония, осложненная ателектазом нижней доли левого легкого и
симптомами сердечно-легочной недостаточности). Имеют место уплотнение и некоторое
увеличение объема икроножных мышц. Сохраняются симптомы кардиомегалии и
гепатомегалии.
37.
В биохимическом анализе крови обнаруживается повышение уровня трансаминаз. Уровень глюкозы 5,3 ммоль/л. В дальнейшем у ребенка постоянноотмечалась гиперфер-ментемия на фоне нормогликемии. Показатель PQ по
электрокардиографическим (ЭКГ) данным — в пределах нормы (0,12 с). В
результате
дополнительного
обследования,
включающего
проведение
электромиографии (ЭМГ), у мальчика были исключены внутриутробные
инфекции, патология мотонейронов (спинальная мышечная атрофия),
митохондриальная патология и врожденные митохондриальные миопатии. В
возрасте 5мес при прогрессивном ухудшении состояния проведено определение
активности фермента а-1,4-глюкозидазы в лаборатории наследственных
болезней обмена веществ (НБО) МГНЦ РАМН, выявившее незначительное
снижение активности данного фермента, что потребовало дальнейшего
проведения ДНК-диагностики с целью уточнения диагноза, впоследствии
подтвердившего БП у ребенка (табл. 3 и 4). В возрасте 5 мес 10 дней ребенок умер
на фоне тяжелой пневмонии, нарастающей сердечнолегочной недостаточности,
инфекционного токсикоза, синдрома диссеминированного внутрисосудистого
свертывания,
микроциркуляторных,
метаболических
и
электролитных
нарушений.
По данным патологоанатомического и гистологического исследования у ребенка
был подтвержден диагноз гликогеноза II типа (БП) с явлениями кардиомегалии
(масса сердца 180 г при норме 36 г), макроглоссии, гепа-томегалии за счет
отложения гликогена в цитоплазме кардиомиоцитов, гепатоцитов, мышцах
языка, пищевода, желудка, диафрагмы.
38.
НаблюдениеМониторинг состояния пациентов с болезнью Помпе
Пациентам с МБП рекомендовано постоянное мониторирование состояния сердечнососудистой, дыхательной, скелетно-мышечной систем, желудочно-кишечного тракта, а
также регулярная оценка качества жизни.
Мониторинг состояния сердечно-сосудистой системы:
- Ребенок с МБП в процессе всего периода лечения нуждается в наблюдении кардиолога,
первоначально в условиях стационара, а при стабилизации состояния - по показаниям,
но не реже одного раза в 3 месяца. Сроки пребывания в стационаре индивидуальны и
зависят от тяжести состояния больного. Уход носит индивидуальный характер.
- В стационаре до стабилизации состояния необходимо обязательно контролировать
ЭКГ (при необходимости - суточное мониторирование ЭКГ с целью своевременного
выявления нарушений ритма сердца). В последующем, при улучшении или
стабилизации состояния, контролировать ЭКГ ежемесячно первые 3 месяца или чаще
по показаниям, далее каждые 3 месяца или по показаниям.
- ЭхоКГ контроль необходим каждые 7-14 дней в первые 12-24 недели ФЗТ (контроль
транзиторного снижения сократимости миокарда, что может сопровождаться
нарастанием проявлений сердечной недостаточности и требует коррекции
симптоматической терапии), далее ежемесячно или по показаниям. По достижении 1
года жизни ЭхоКГ - 1 раз в 3 месяца или по показаниям.
- Контроль NT-proBNP необходим каждые 3 месяца или по показаниям до 1 года, далее
после первого года жизни - 1 раз в 3-6 месяцев или по показаниям.
- Строгий ежедневный контроль объема потребляемой жидкости (питание,
парентеральная инфузионная терапия) с целью предупреждения перегрузки или
обезвоживания (ограничение жидкости до 2/3 от физиологической потребности).
39.
Мониторинг состояния дыхательной системы:- Во время каждого осмотра пациента необходимо оценивать респираторный статус (контроль характера и частоты дыхания
при бодрствовании и во сне, ведение родителями респираторного дневника).
- Оценка легочной функции и газообмена должна проводиться при постановке диагноза, каждом визите пациента, при изменении
клинического состояния (при присоединении вирусной инфекции). Рентгенограмма грудной клетки должна проводиться при
постановке диагноза и по показаниям.
- Полисомнография - после постановки диагноза и по клиническим показаниям (выявление и профилактика апноэ во сне).
- Микробиологическое исследование трахеального аспирата с целью оценки микробного статуса и необходимости
проведения антибактериальной и/или противогрибковой терапии.
- Оценка ФВД во сне и/или при бодрствовании - при появлении и/или нарастании жалоб на усталость, появление
периферического цианоза, апноэ во сне, падении ЖЕЛ ниже 40-50%, падении сатурации и неэффективности ее
коррекции на фоне инсуфляций кислорода у детей раннего возраста.
- Неинвазивная кислородная поддержка (создание положительного давления) - при развитии гипоксемии,
обструктивном синдроме.
- Лечение любых инфекций дыхательных путей должно носить агрессивный характер, назначение антибактериальной
терапии проводится в максимальных терапевтических дозировках.
- Обучение родителей использованию небулайзера, отсоса.
Мониторинг состояния желудочно-кишечного тракта:
- Контроль росто-весовых параметров еженедельно, ежемесячно, ежеквартально
- Включение в рацион специализированных высококалорийных и высокобелковых, анитрефлюксных смесей.
- Зондовое кормление, или совмещение перорального и зондового питания при сохранении адекватного глотания и
отсутствии поперхивания.
- Назодуоденальное зондовое кормление при сохранении рефлюкса и высоком риске аспирации.
- Оральная стимуляция, сосание соски, гимнастика должны проводиться всем пациентам для поддержания
нормального развития навыков глотания и устной речи.
Мониторинг состояния скелетно-мышечной системы:
- Поддержка максимального уровня функций скелетно-мышечного аппарата для предотвращения и минимизации вторичных
осложнений (развитие контрактур, мышечных атрофий, компенсаторной деформации скелета, остеопении, остеопороза).
- При проведении реабилитационных мероприятий избегать чрезмерных (максимальных) нагрузок. Регулярно:
физиотерапевтические процедуры, массаж, ЛФК, логопедия, респираторная терапия (дыхательная гимнастика), обучение
самообслуживанию и использованию вспомогательных приспособлений (зависит от возраста ребенка). По показаниям занятия с логопедом, сурдологом.
- Осмотр невролога с оценкой моторных возможностей и нервно–психического развития ребенка необходимо проводить каждые
3-6 месяцев для детей в возрасте до 5 лет и ежегодно для более старших пациентов (оценка по шкале Альберта моторного
развития младенцев - в Приложении Г3). Тестирование слуха должно проводиться ежегодно или по клиническим показаниям.
40.
Пациентам с БППН рекомендовано постоянное мониторирование состояния дыхательной,скелетно-мышечной систем, а также регулярная оценка качества жизни.
Пациенты с БППН должны проходить обязательные ежегодные обследования минеральной
плотности костей для своевременной коррекции получаемого лечения по поводу остеопении и
остеопороза. Следует настойчиво рекомендовать пациенту с нарушением походки и
равновесия пользоваться вспомогательными предметами - тростью, ходунками, а при
необходимости использовать механические средства передвижения (кресло-каталку и пр.).
Пациенты с БППН должны регулярно осматриваться эндокринологом с проведением
обследования. Большое значение имеет правильный образ жизни с адекватным распределением
нагрузок, занятий лечебной физкультурой под контролем информированного инструктора.
Для мониторинга состояния пациентов, находящихся на патогенетической терапии,
рекомендовано их наблюдение не реже одного раза в год в специализированных центрах, в
которых может быть оказан объем медицинской помощи в соответствии с данными клиническим
рекомендациями.
41.
Список используемой литературы:Основная литература:
Клинические рекомендации/ Болезнь Помпе 2017 г.
Клинические рекомендации/ Болезнь Нимана-Пика 2015 г.
Дополнительные источники:
• Клиническая генетика : учебник / Н. П. Бочков, В. П. Пузырев,С. А. Смирнихина, 2018
• Клиническая генетика: учебник/ В.И. Иванов - изд., испр. и доп. - М., 2011.
• Herzog A., Hartung R., Mengel E. et al. Genotype-phenotype correlations in Pompe disease/ 2011.
• http://onevroze.ru/tipy-bolezni-nimana-pika-diagnostika-lechenie-i-dieta-pri-etomzabolevanii.html
• https://www.mediasphera.ru/issues/zhurnal-nevrologii-i-psikhiatrii-im-s-s-korsakova2/2017/11/1199772982017112062
• https://cyberleninka.ru/article/n/mladencheskaya-forma-bolezni-pompe-klinika-diagnostika-ilechenie