



")













")







 medicine
medicineSimilar presentations:





")




Редкие заболевания с умственной отсталостью
1. Редкие заболевания с умственной отсталостью
Специальная психологияТольяттинский Государственный
Университет
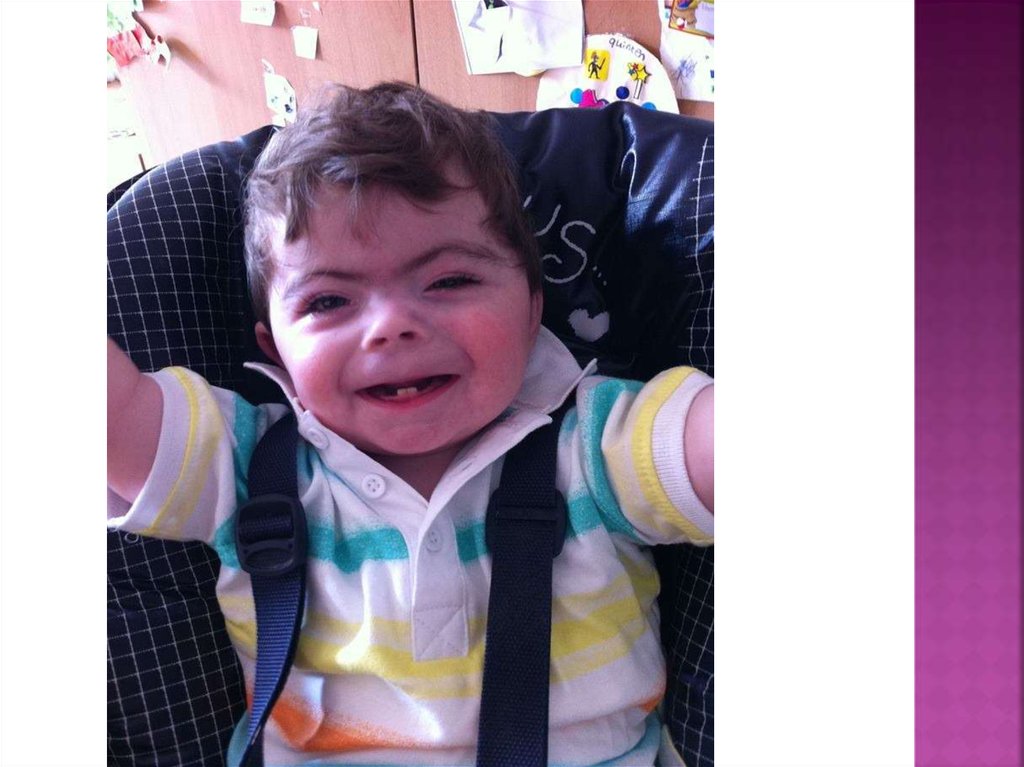
2. Синдром корнелии де ланге
СиндромКорнелии де Ланге (синдром
Брахмана-Ланге) — наследственное
заболевание, проявляющееся умственной
отсталостью и множественными
аномалиями развития. Частота
заболевания — примерно 1 на 10000.
Впервые случай такого заболевания описан
в 1916 году немецким врачом
В.Брахманом, название синдром получил
по имени голландского педиатра Корнелии
де Ланге, в 1933 году описавшей синдром
на основе анализа пяти случаев
заболевания.
3.
4.
5. Клиническая картина синдрома корнелии де ланге
Микроцефалия (уменьшение размеров черепа более чем на10 % возрастной нормы);
Брахицефалия (укорочение черепа в сагиттальном
направлении, в результате чего поперечный размер головы
увеличивается, а продольный уменьшается);
Тонкие сросшиеся брови; длинные загнутые ресницы;
деформированные ушные раковины; маленький нос, открытые
вперед ноздри, атрезия хоан; тонкая верхняя губа;
Микрогения; высокое нёбо или расщелина нёба; нарушение
прорезывания зубов;
Миопия, косоглазие, астигматизм, атрофия зрительных
нервов, колобома зрительного нерва;
Маленькие кисти и стопы, отсутствие или значительное
недоразвитие проксимальных отделов конечностей,
вследствие чего кисти и стопы кажутся прикрепленными
непосредственно к туловищу, уменьшение количества
пальцев;
6. Клиническая картина синдрома корнелии де ланге
Мраморная кожа;Гипоплазия сосков;
Гипертрихоз;
Судороги;
Врожденные пороки внутренних органов (сердца, почек,
пилоростеноз, крипторхизм) и др.
У всех больных отмечаются отставание в росте, глубокая
умственная отсталость; типичны рецидивирующие
респираторные инфекции.
Выделяют два варианта синдрома:
первый (классический) с выраженной пренатальной
гипоплазией, значительной задержкой физического и
интеллектуального развития, грубыми пороками развития;
второй — с аналогичными лицевыми и малыми скелетными
аномалиями, но пограничной задержкой психомоторного
развития и отсутствием грубых пороков развития.
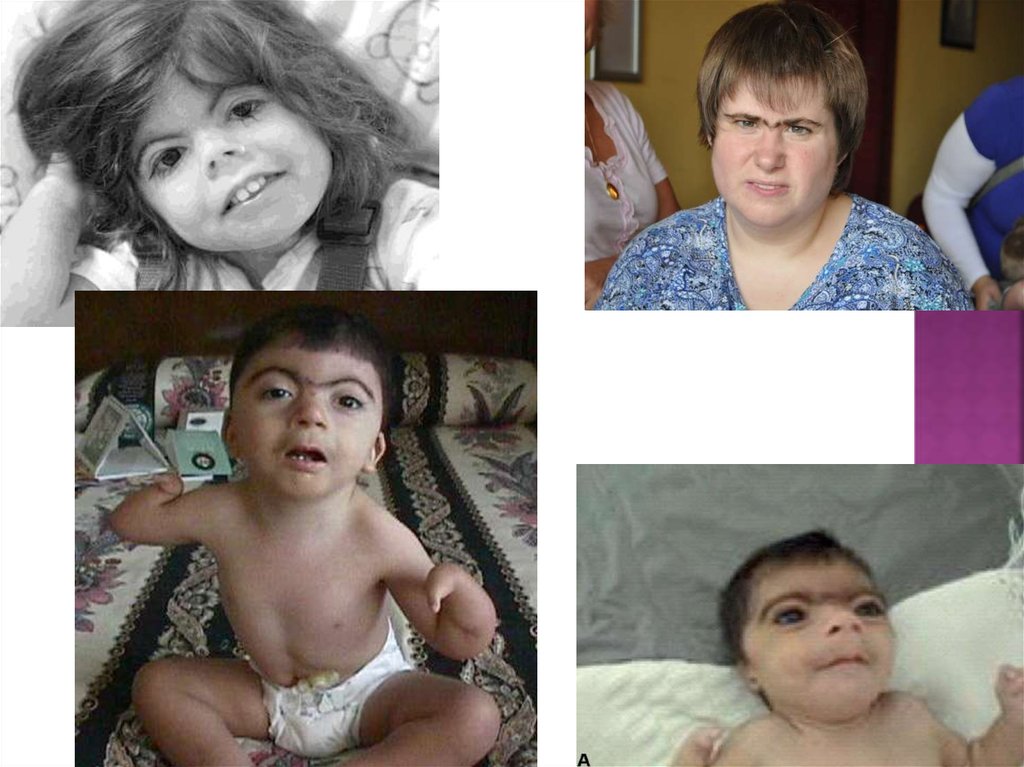
7. Синдром уильямса (синдром эльфа)
Синдром Уильямса (синдром «лица эльфа») —синдром, возникающий как следствие
наследственной хромосомной перестройки,
страдающие которым обладают
специфической внешностью и
характеризуются общей задержкой
умственного развития при развитости
некоторых областей интеллекта.
Синдром описан в 1961 году кардиологом из
Новой Зеландии Дж. Уильямсом, который
выделил среди своих пациентов, у которых
были обнаружены сходные дефекты сердечнососудистой системы, людей, которые также
имели схожую внешность и умеренную
отсталость.
8. Особенности внешности
Больные имеют особое строение лица, в специальной литературеназываемое «лицом эльфа», поскольку оно напоминает лицо эльфов в
их традиционном, фольклорном варианте. Для них характерны
широкий лоб, разлёт бровей по средней линии, опущенные вниз
полные щёки, большой рот с полными губами (особенно нижней),
плоское переносье, своеобразная форма носа с плоским тупым
концом, маленький, несколько заострённый подбородок.
Глаза зачастую ярко-голубые, со звёздчатой картиной радужки и
склерами синеватого цвета. Разрез глаз своеобразный, с
припухлостями вокруг век. Сходящееся косоглазие.
Для старших детей характерны длинные, редкие зубы. С возрастом
лицо больных несколько меняется: появляется массивность
надбровных дуг, меньше выражена пастозность лица, нет плоского
переносья и эпиканта. Обращает на себя внимание увеличенное
расстояние от основания носа до верхней губы.
Сходство лиц усиливает улыбка, которая ещё больше подчёркивает
отёчность век и своеобразное строение рта.
Ни одна из этих черт не является обязательной, но их общее
сочетание всегда присутствует.
9.
10.
Для этого синдрома характерен дефицит нагляднообразного мышления. Степень интеллектуальногодефекта обычно довольно значительна, однако бывают
случаи более легкой интеллектуальной
недостаточности, средние IQ = 56 с колебаниями от 40
до 80.
Можно отметить большое сходство психопатологической
картины дефекта у всех больных: при значительном
снижении интеллекта речь у детей довольно хорошая,
больные имеют относительно большой словарный запас,
очень словоохотливы, склонны к подражанию. Вместе с
тем всегда страдают пространственные представления,
организация и планирование деятельности. Очень
характерны и постоянны особенности личности этих
детей: добродушие, приветливость, послушание.
Практически всегда имеется хороший музыкальный слух
даже при выраженном интеллектуальном дефекте.
Нередко выявляются неврозоподобные нарушения —
энурез, страхи, навязчивые действия.
Некоторые дети могут учиться во вспомогательной
школе, они овладевают чтением и письмом, но им
недоступны действия, связанные с организацией даже
простейших трудовых операций.
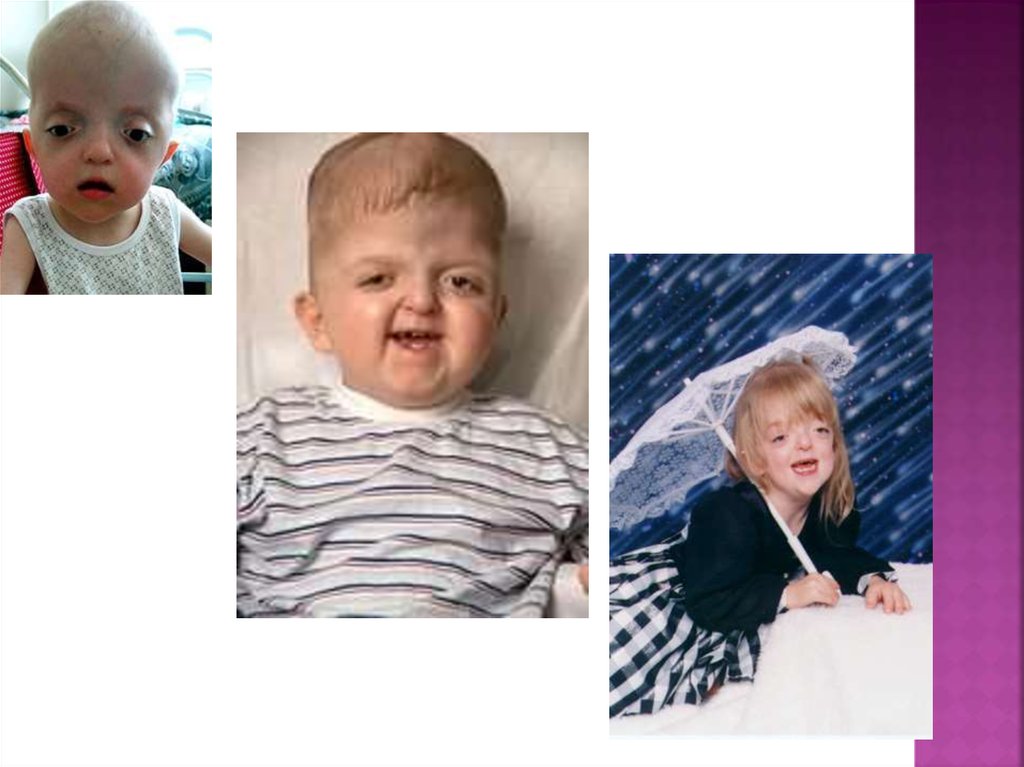
11. Синдром аперта
Синдром Апера (Аперта)(акрокраниодисфалангия,
акросфеносиндактилия,
акроцефалосиндактилия) — врожденная
аномалия развития черепа, которая сочетается
с отклонением развития кистей рук. Раннее
закрытие венечного и стреловидного швов
способствует деформации черепа, что
приводит к внутричерепной гипертензии.
Синдром Апера является одной из форм
акроцефалосиндактилии.
Впервые это заболевание описал французский
педиатр Эжен Аперт в 1906 году. Заболевание
встречается у одного новорожденного на 160
000—200 000.
12.
13.
Однимиз признаков заболевания является
то, что краниосиностоз сочетается с
брахикефалией. В связи с
преждевременным срастанием
корональных швов увеличивается
внутричерепное давление, что обычно
приводит к умственной отсталости.
Немаловажными признаками синдрома
являются высокий, выпуклый лоб, плоское
или вогнутое лицо, в результате чего
наблюдается нарушение костей лицевого
черепа, что приводит к деформации
челюсти, а также синдактилия рук и ног.
14. Синдактилия двух пальцев у младенца
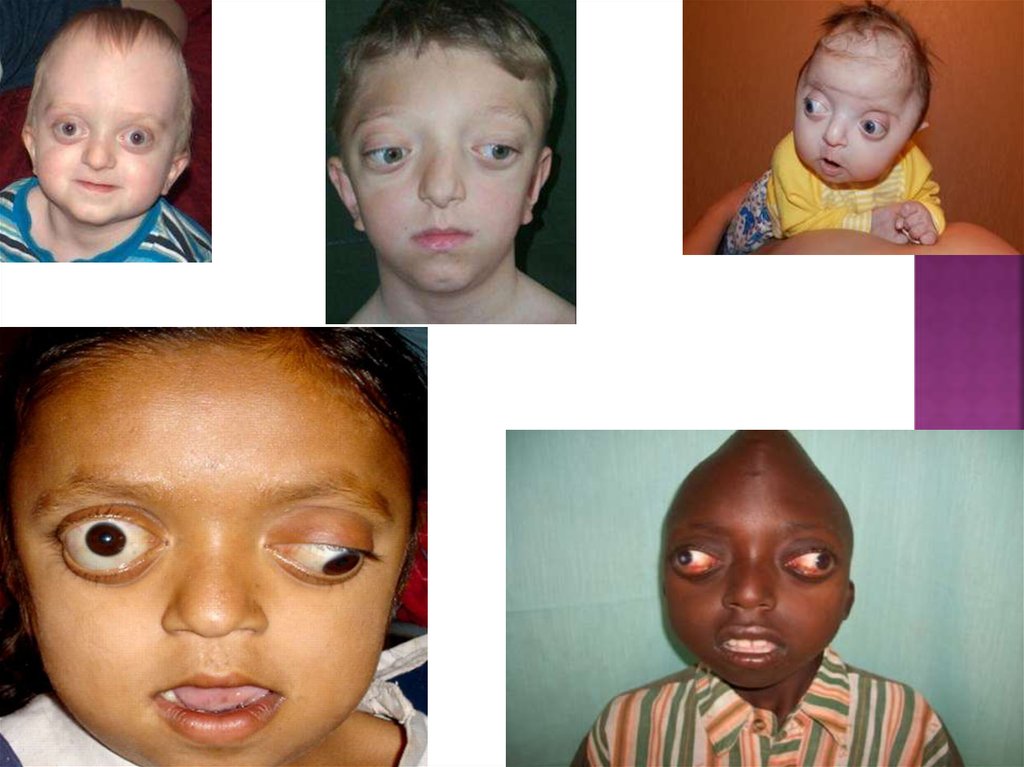
15. Синдром крузона
Синдром Крузона являетсягенетическим заболеванием,
приводящим к ненормальному
сращиванию костей черепа и
лица.
Младенцы имеют швы между
костями лица и черепа. Когда
мозг ребенка растет, эти швы
позволяют черепу расширяться.
Швы полностью зарастают
когда мозг полностью
сформирован и его рост
останавливаетя.
При синдроме Крузона кости
черепа и лица срастаются
слишком рано, и череп затем
вынужден расти в направлении
оставшихся открытыми швов.
Это приводит к аномальной
форме головы, лица и зубов.
16.
17.
Причины синдрома КрузонаСиндром Крузона вызван генетическим
дефектом. Пока не ясно, какие причины
заставляют ген, вызывающий синдром
Крузона, мутировать. Некоторые из
мутировавших генов могут быть унаследованы
от генов родителей.
Факторы риска синдрома Крузона
Факторы, которые могут увеличить риск
появления синдрома Крузона у ребенка,
включают:
Родители с синдромом Крузона;
Родители, которые не имеют данного
расстройства, но которые имеют ген,
вызывающий синдрома Крузона;
Взрослый или пожилой возраст отца на момент
зачатия.
18. Симптомы синдрома Крузона
Уплощенная верхняя и задняя части головы;Уплощенной лоб и виски;
Средняя часть лица имеет небольшой размер и вытянута больше, чем
обычно;
Остроконечный нос;
Сжатые носовые проходы, что часто приводит к уменьшению потока
воздуха через нос;
Большая, выдающаяся вперед нижняя челюсть;
Смещение зубов;
Узкое небо, или волчья пасть.
Другие симптомы и осложнения, которые могут возникнуть в
результате синдрома Крузона включают в себя:
Проблемы с развитием внутреннего уха и потеря слуха;
Болезнь Меньера - головокружение или звон в ушах;
Проблемы с глазами, в том числе проблемы со зрением, косоглазие
или непроизвольные движения глаз;
Искривление позвоночника;
Головные боли;
Акантоз - маленькие, темные, бархатистые пятна на коже;
Гидроцефалия - накопление жидкости в черепе.
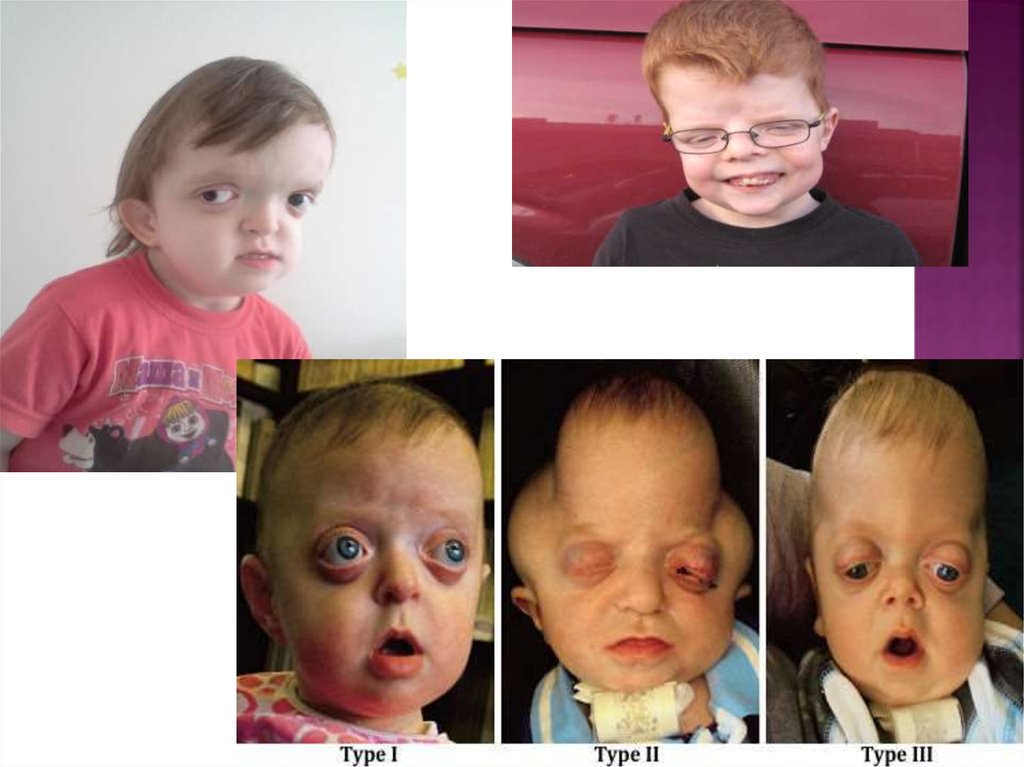
19. Синдром Пфайффера
- это редкое генетическое расстройство,характеризующееся преждевременным
"срастанием" некоторых костей черепа
(краниостеноз), которое препятствует
дальнейшему росту черепа и тем самым
повреждает форму головы и лица. Синдром
Пфайффера также влияет на кости рук и ног.
Данный синдром назван в честь Рудольфа Артура
Пфайффера (род.1931),который в 1964 году
перечислил черты лица, которые присущи кроне
синостоза, такие как высокий выпуклый лоб и
верхнечелюстная гипоплазия (значительно
выпирающие глаза, подпирающие кости щёк). С
Синдромом Пфайффера рождается один ребёнок
на 100 000 новорождённых.
20.
21.
22.
Многиеособенности черт лица, которые
описывает Пфайффер являются
результатом преждевременного
"срастания" костей черепа. Голова не
способна нормально расти и развиваться,
что ведёт к тому, что глаза приобретают
выпуклую форму и становятся широко
раскрытыми, верхняя челюсть
слаборазвита, а нос выглядит как клюв.
Около 50 процентов детей с синдромом
Пфайффера теряют слух и имеют
проблемы с зубами. Широкие большие
пальцы на руках и ногах - особые признаки
этого синдрома.
23.
Синдром Пфайффера подразделяется на тритипа.
1 тип, также известен как классический
синдром Пфайффера, имеет симптомы,
перечисленные выше. Большинство людей с
этим типом имеют достаточно полноценную
жизнь и владеют нормальным интеллектом.
2 и 3 типы более серьёзные и связаны с
проблемами нервной системы.
Преждевременное "срастание" костей черепа
может ограничить рост мозга, что может
привести к задержке в развитии и другим
неврологическим проблемам.
2 тип отличается от 3 типа тем, что может привести
к формированию головы в виде клеверного листа,
причиной этого является обширное "срастание"
костей черепа.
24. Синдром Тричера Коллинза (синдром франческетти )
Синдром Тричера Коллинза (англ. TreacherCollins syndrome, TCS, челюстно-лицевой
дизостоз) — аутосомно-доминантное
заболевание, характеризующееся черепнолицевой деформацией. Описан английским
офтальмологом Эдвардом Тричером
Коллинзом в 1900 году.
Синдром Тричера Коллинза встречается у 1 из
50 000 младенцев. Типичные клинические
признаки: грубый дефект лицевой части
черепа, косоглазие, колобомы век; размер
рта, подбородка и ушей существенно меньше
нормы. В некоторых случаях — ослабление
слуха.
25.
26.
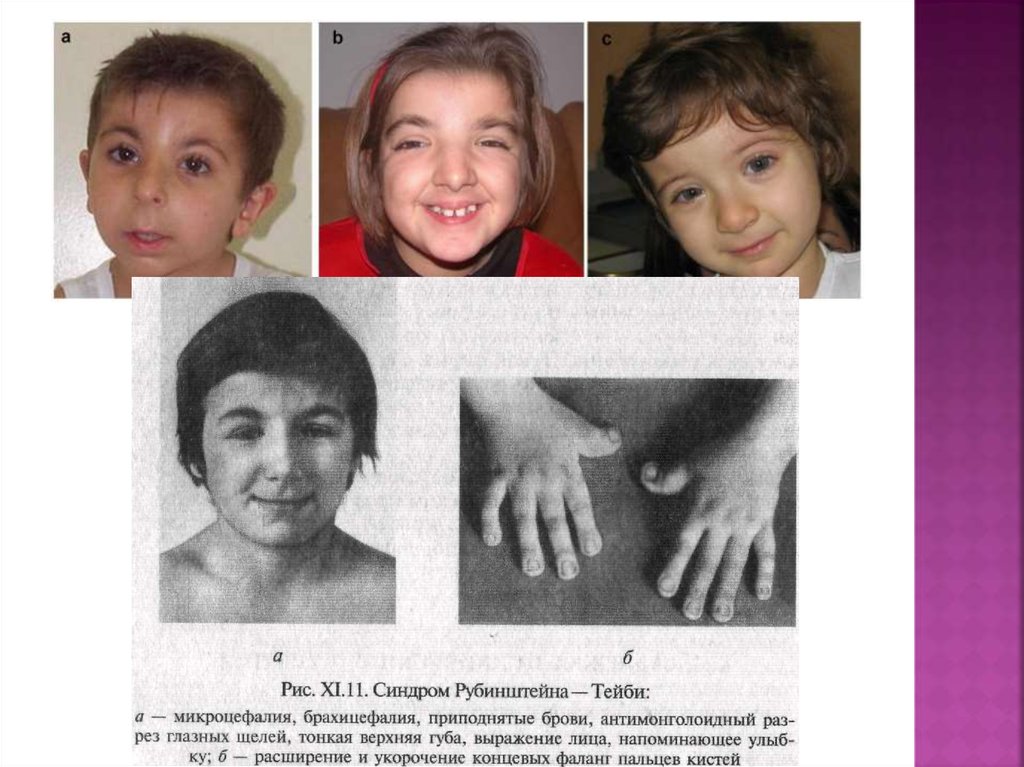
27. Синдром рубинштейна – тейби
Синдром Рубинштейна-Тейби (синоним: синдромширокого I пальца кистей и стоп, специфического лица
и умственной отсталости) впервые описан в 1963 г.
Популяционная частота — 1:25 000 — 30000,
соотношение полов — 1:1.
Тип наследования аутосомно-доминантный (ген CREBBP
в локусе 16p13.3), но в большинстве случаев мутация
возникает спонтанно, то есть не наследуется от
родителей.
Дифференциальный диагноз: брахидактилия, тип D,
акроцефалосиндактилия, тип VI.
Минимальные диагностические признаки:
прогрессирующая умственная отсталость, широкие
терминальные фаланги первых пальцев кистей и стоп,
характерное лицо, отставание роста и костного
возраста, микроцефалия, крипторхизм.
28.
29.
Основными признаками синдрома являются отставаниепсихофизического развития, дисплазия лица, аномалии
развития пальцев. У 94 % больных рост ниже среднего.
Костный возраст значительно отстает от паспортного
(94 %). Отмечается умственная отсталость (100 %) (в
большинстве случаев — глубокая олигофрения,
задержка моторного и речевого развития).
Комплекс черепно-лицевых дисморфий включает
брахицефалию, микроцефалию, большой и поздно
закрывающийся родничок, выступающий лоб с низким
ростом волос, приподнятые дугообразные брови,
антимонголоидный разрез глаз (93 %), широкую
переносицу, эпикант (62 %), длинные ресницы, птоз,
широкую спинку носа (71 %), загнутый книзу кончик носа
(клювовидный нос) (90 %), гипоплазию крыльев носа
(72 %), умеренную ретрогнатию, гримасу,
напоминающую улыбку, высокое арковидное небо
(93 %).
Со стороны глаз отмечаются косоглазие (79 %) и
аномалии рефракции (58 %). Ушные раковины
деформированы, уменьшены или увеличены (74 %).
30.
Аномалии пальцев заключаются в расширении,укорочении и уплощении ногтевых фаланг первых
пальцев кистей и стоп (100 %), иногда терминальных
фаланг других пальцев кисти (60 %), вальгусной
деформации межфаланговых суставов, удвоении
ногтевой фаланги первых пальцев стоп (30 %), реже —
проксимальной фаланги, в ряде случаев — полидактилии
стоп, частичной синдактилии кистей и стоп.
Отмечаются также лордоз, кифоз, сколиоз, аномалии
грудины и рёбер, уплощение крыльев тазовых костей.
В половине случаев встречаются гирсутизм, яркокрасный невус на коже лба, затылка, боковой
поверхности шеи. Часты изменения дерматоглифических
показателей.
Пороки развития внутренних органов включают
незаращение артериального протока и дефекты
перегородок сердца, одностороннюю аплазию почек,
удвоение почек, гидронефроз, расширение
мочеточников или их стеноз, дивертикул мочевого
пузыря, крипторхизм (в 79 % случаев), нарушение
лобуляции легких, агенезию или аплазию мозолистого
тела(обнаруживаемую при пневмоэнцефалографии или
на вскрытии).
31. Другие редкие синдромы с у.о.
Болезнь Тея – СаксаСиндром Аспергера
Синдром Патау *
Синдром Шейе (синдром Гурлера - гаргоилизм)
Синдром Нунан
Синдром Шерешевского – Тёрнера **
Синдром Лея
Синдром Ретта
Синдром Ангельмана
Синдром Эдвардса *
Синдром Клайнфельтера *
Синдром Лежена (синдром кошачьего крика)*
32. Обязательно прочитать!!!
Г.С.МАРИНИЧЕВА, М.Ш. ВРОНО.
УМСТВЕННАЯ ОТСТАЛОСТЬ.
Глава
2.2. Наследственные формы
умственной отсталости. Синдромы с
множественными врожденными
аномалиями.