




















































 medicine
medicine biology
biologySimilar presentations:










Хромосомные болезни
1.
Хромосомныеболезни
Илатовская С.А., преподаватель генетики
СПб ГБПОУ «Акушерский колледж»
2.
Хромосомные болезниПричина - хромосомные или геномные мутации
В настоящее время насчитывают почти 1000
хромосомных болезней
Основные проявления – летальность и врожденные
пороки развития. Патология начинает проявляться со
стадии зиготы и является одной из главных причин
внутриутробной гибели:
30-40% зигот погибает от хромосомных или геномных
мутаций до имплантации;
У 2-4 недельных абортусов (эмбрион с оболочками)
хромосомные и геномные аномалии
обнаруживаются в 60-70% случаев.
3.
Анеуплоидия аутосомВ большинстве случаев
дополнительная хромосома
имеет материнское
происхождение. Частота
зависит от возраста матери
4.
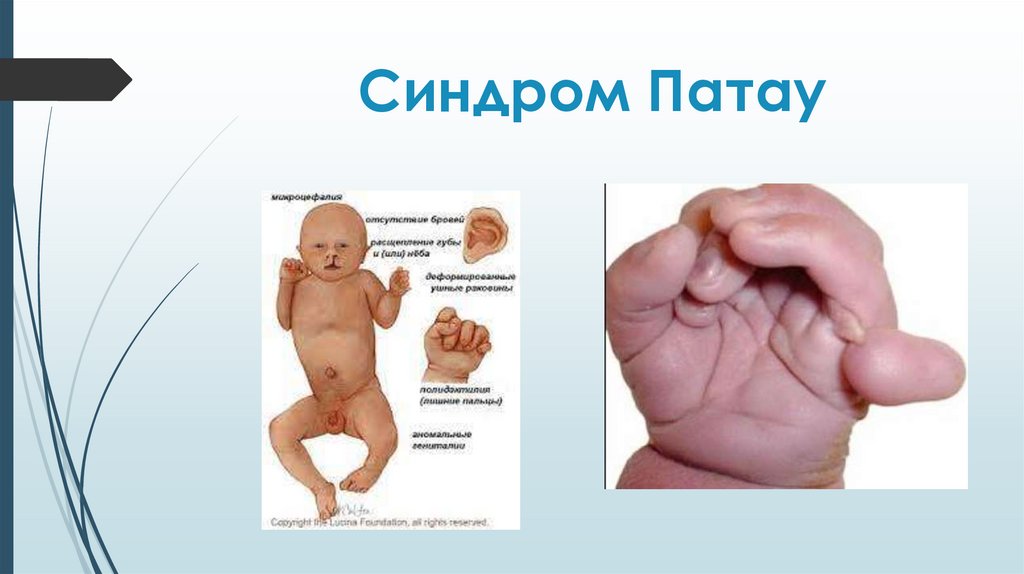
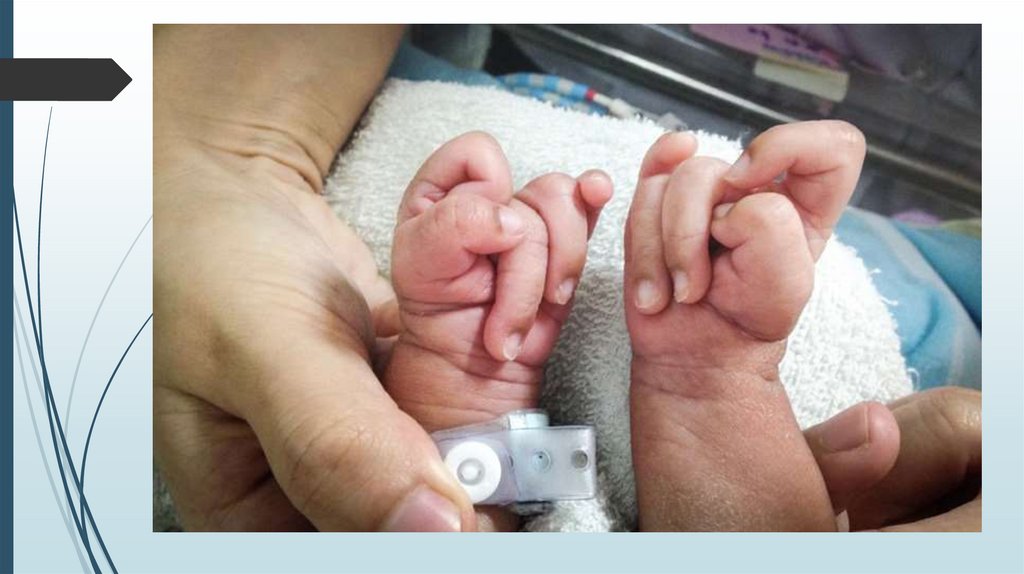
Синдром ПатауТрисомия по 13 паре хромосом.
Кариотип 47, ХХ +13; 47, ХУ +13.
Основные причины:
нерасхождение хромосом;
робертсоновская транслокация.
Частота 1:15 000 – 1:25 000
5.
Синдром Патау6.
Синдром Патау7.
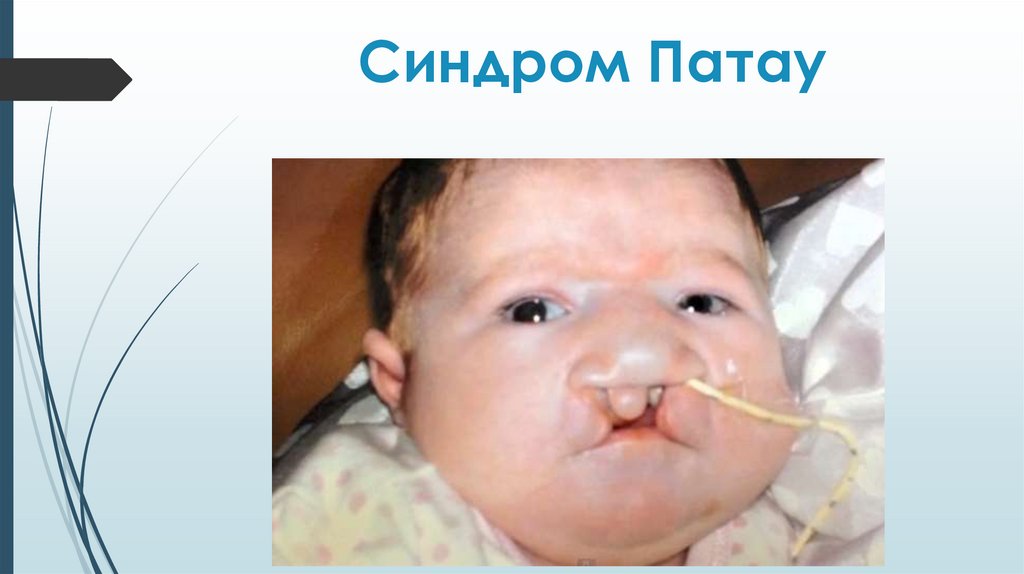
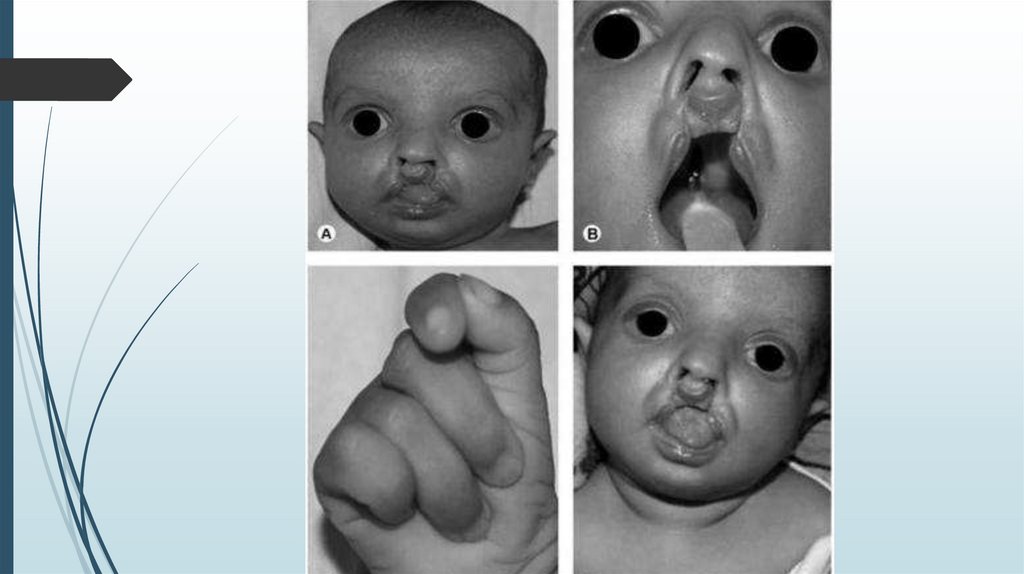
Синдром ПатауМножественные врожденные пороки развития
головного мозга и лица. Микроцефалия.
В теменной области - участок отсутствия кожи до
1 см в диаметре. Лоб скошенный, глазные щели
узкие, переносица запавшая, глаза недоразвиты
с помутнением роговицы, уши расположены
низко и деформированы. Типичный признак
синдрома Патау - двусторонние расщелины
верхней губы и неба.
8.
Синдром ПатауДвусторонняя полидактилия на руках,
флексорное положение кистей, дефекты
перегородок сердца, незавершенный
поворот кишечника, дефекты
поджелудочной железы и печени. Почки
увеличены в размерах, поликистоз.
Неопущение яичек (крипторхизм) и
недоразвитие полового члена у мальчиков,
удвоение матки и влагалища у девочек.
9.
Синдром ПатауЛицо: низко расположены ушные раковины; расщелина
верхней губы и неба; микроцефалия; анофтальмия,
микроофтальмия; катаракта.
Нервная система: аплазия мозолистого тела;
аринэнцефалия, голопроэнцефалия (неразделение двух
полушарий); гипоплазия мозжечка, черепа, зрительных
трактов.
Костно-мышечные и кожные проявления: полидактилия,
аномальный вид кисти; выступающая пятка, стопа-качалка;
омфалоцеле (пупочная грыжа); врожденное отсутствие
участков кожи и волос.
Врожденные пороки сердца, одна пуповинная артерия.
10.
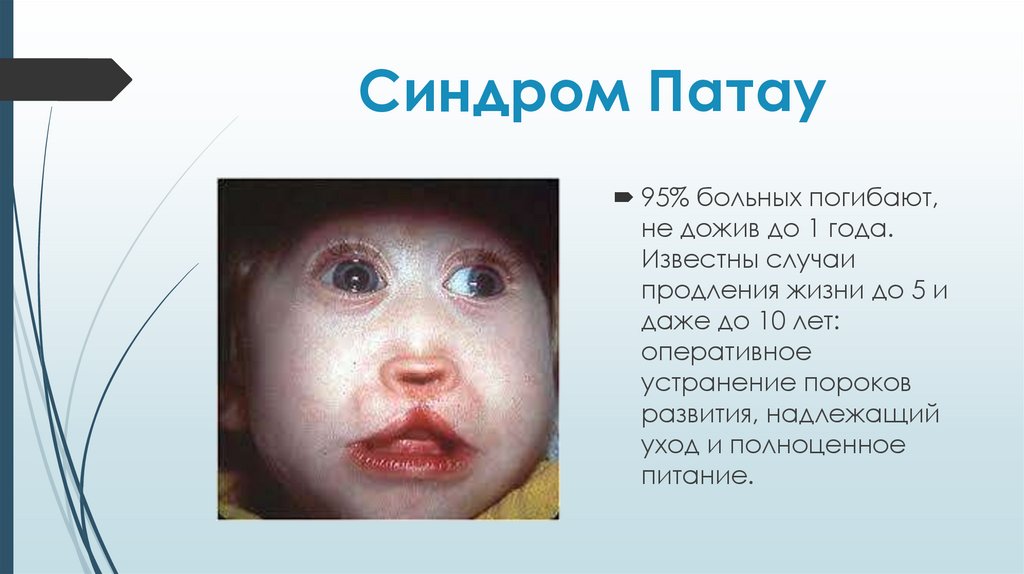
Синдром Патау95% больных погибают,
не дожив до 1 года.
Известны случаи
продления жизни до 5 и
даже до 10 лет:
оперативное
устранение пороков
развития, надлежащий
уход и полноценное
питание.
11.
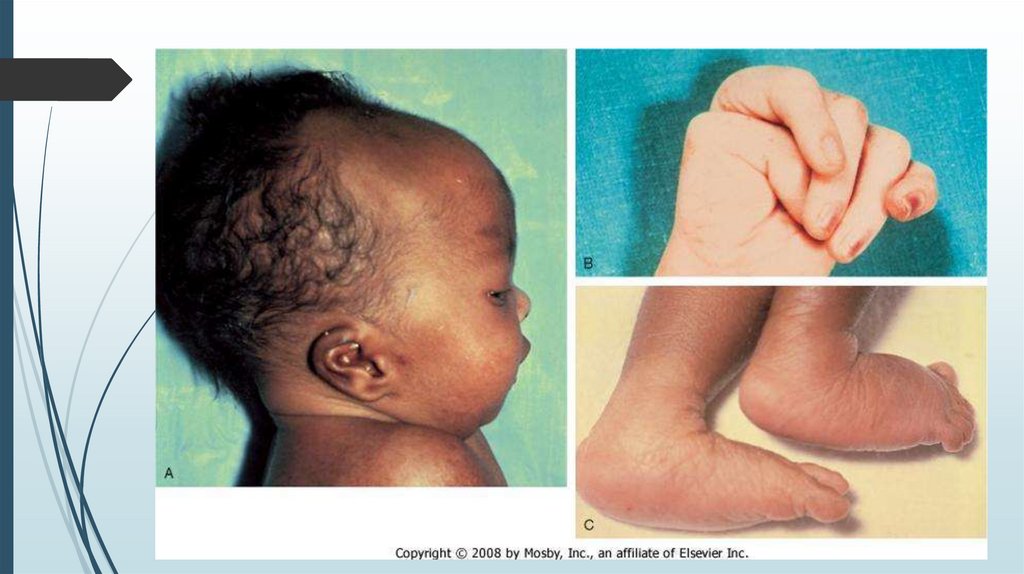
Синдром ЭдвардсаВторое по частоте встречаемости
хромосомное заболевание после синдрома
Дауна. Частота 1:5000-1:7000. Среди девочек
встречается в 3 раза чаще, чем среди
мальчиков. Описан в 1860 году педиатром
Джоном Эдвардсом.
Трисомия по 18 паре хромосом. Кариотип
47, ХХ +18; 47 ХУ +18; 10% - мозаичные формы
12.
13.
Синдром ЭдвардсаФакторы риска:
1.Возраст матери – после 35 лет
возрастает с каждым годом
2.Прием лекарственных средств,
способных влиять на деление клеток
3.Курение и алкоголь
14.
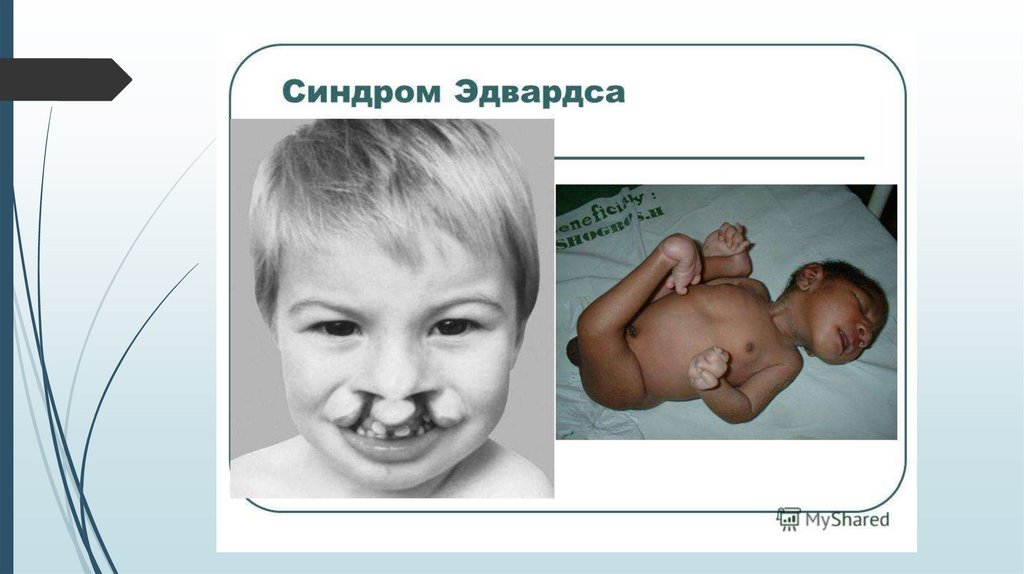
СимптомыНизкая масса тела при рождении – до 2 кг
Нарушение формы черепа: долихоцефалия
(длинноголовость), микроцефалия, гидроцефалия, узкий лоб
Перекрещивание пальцев кистей, сжатые кулачки,
флексорное положение кистей
Мелкие черты лица, короткие глазные щели, катаракта
Маленький рот и нижняя челюсть (микрогения)
Деформированные ушные раковины, сужение или
отсутствие наружного слухового прохода
Дисплазия стоп, кистей, вывих т/б сустава
Пороки сердца, ЖКТ, анального отверстия, мочеполовой
системы.
15.
16.
17.
Синдром Эдвардса18.
Синдром Эдвардса60% детей доживают до 3 месяцев; до 1
года 5-10%
Основная причина смерти – остановка
дыхания и нарушения работы сердца и
почек.
Оставшиеся в живых глубокие
олигофрены
19.
Синдром Дауна20.
Синдром ДаунаЧастота встречаемости 1:600-1:800
Кариотип 47, ХХ +21; 47, ХУ +21 – 94%;
4% - результат транслокации;
2% - мозаичные формы.
Впервые описал английский врач Джон Даун в
1866 году.
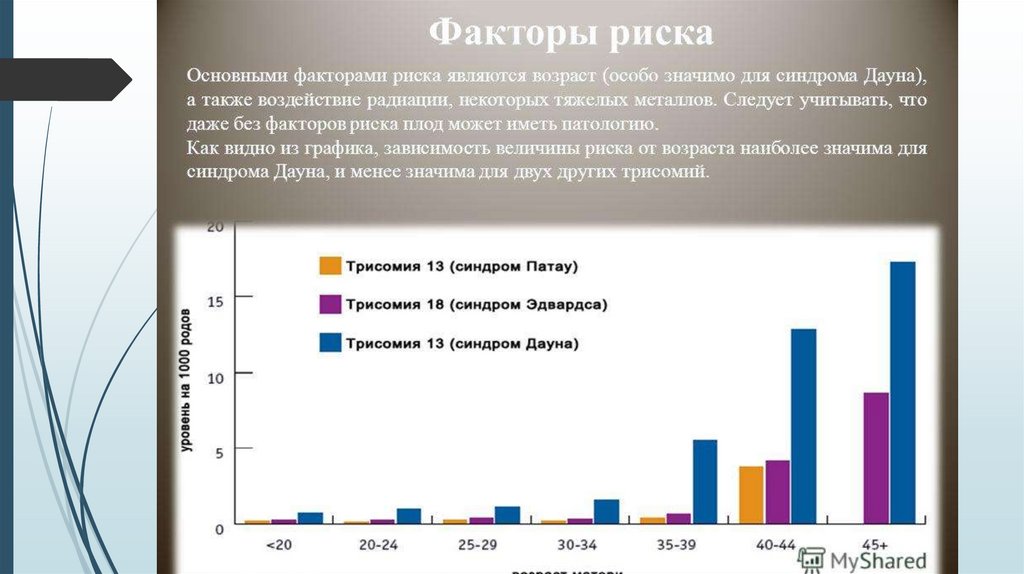
Достоверно установленным фактором риска
является возраст родителей: частота у матерей
старше 35 лет составляет 1:35. Возраст отца
старше 42-45 лет также может послужить
причиной развития синдрома.
21.
22.
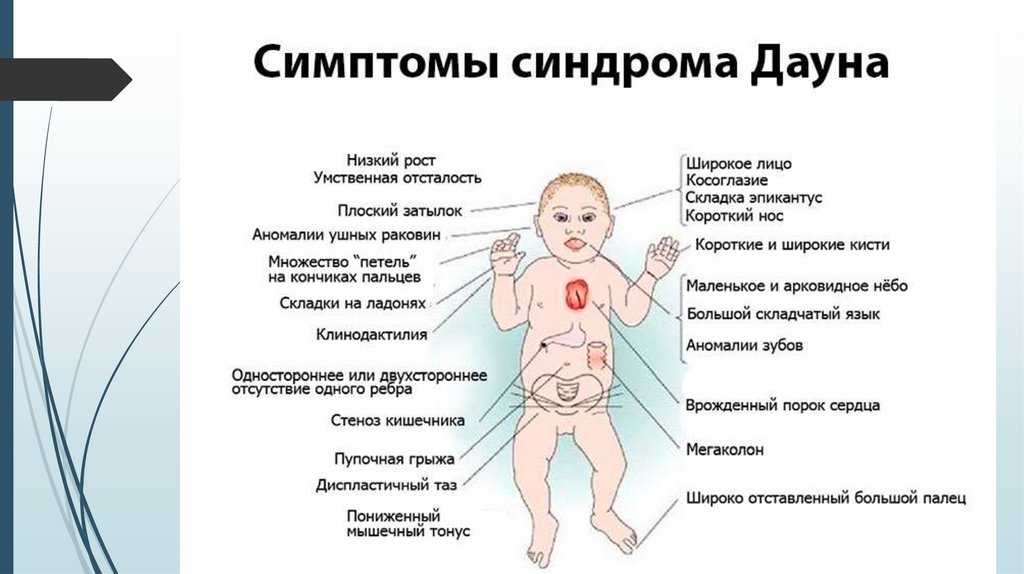
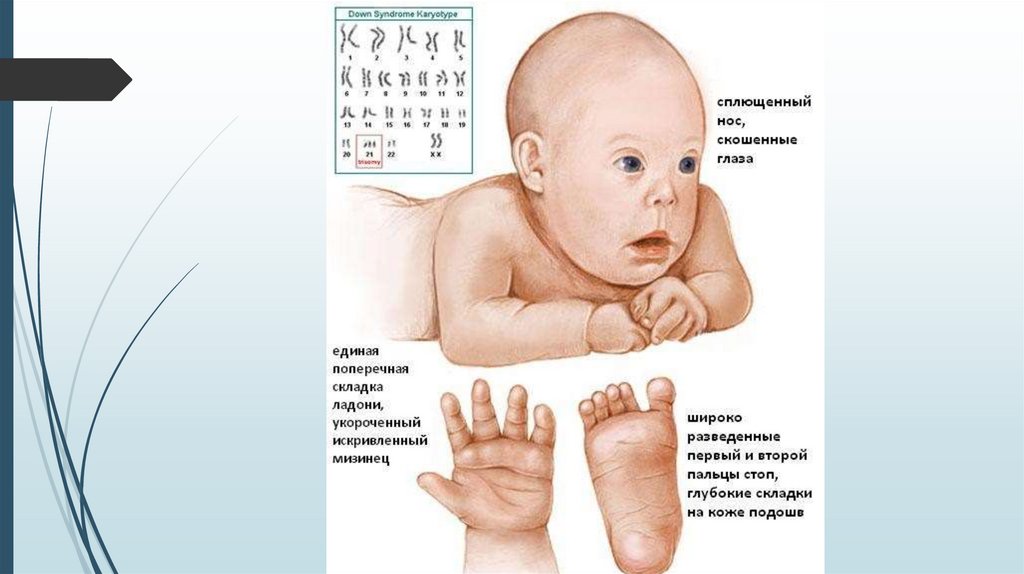
Фенотип синдрома Даунаголова с уплощенным затылком;
толстая кожная складка на задней поверхности
шеи;
скошенный и узкий лоб;
лицо плоское, переносица широкая и
вдавленная;
язык большой и виден между губами;
постоянно открытый рот, толстые губы;
«монголоидный» разрез глаз; эпикант;
ушные раковины маленькие и
деформированные.
23.
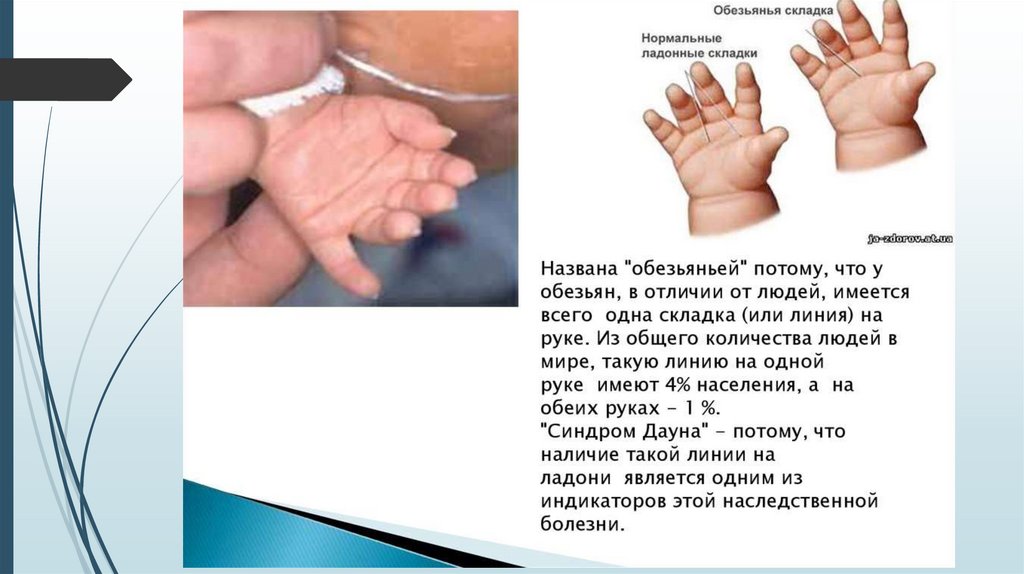
Костно-мышечная системанизкий рост;
короткая шея;
воронкообразная или килевидная грудная клетка;
широкие кисти и стопы с короткими пальцами;
поперечная борозда на ладонях;
первый палец на стопах широко отстоит от
других пальцев - «сандалевидная щель»;
мышечная гипотония с разболтанностью
суставов, поэтому новорожденные с синдромом
Дауна лежат в кроватке, раскинув ручки и ножки.
24.
ВПР при синдроме Даунау 50% больных - врожденные пороки
сердца (дефекты межпредсердной
или межжелудочковой
перегородок), пищеварительный
тракт поражается в 15% случаев
(атрезии или стенозы
двенадцатиперстной кишки).
25.
Умственная отсталостьдебильность - в 75% случаев,
имбецильность - у 20% больных,
идиотия - в 5% случаев.
Дети легче осваивают навыки,
связанные с физическими
движениями, чем речевые. Дети
внимательные, ласковые, терпеливые
при обучении.
26.
27.
28.
29.
30.
Синдромычастичных
анеуплоидий
31.
Синдром «Кошачьего крика»32.
Синдром кошачьего крикаЧастичная моносомия
короткого плеча 5 хромосомы
(5p-), обусловлен делецией.
Открытие Дж. Лежена в 1963 г.
Частота 1:45 000
33.
Синдром кошачьего крикаЦитогенетическое исследование. Чаще всего это делеция
от 1/3 до ½ длины короткого плеча 5 хромосомы. Потеря
всего короткого плеча встречается редко.
Для развития клинической картины имеет значение не
величина утраченного участка, а конкретный фрагмент
хромосомы.
За развитие полного синдрома отвечает незначительный
участок - 5p15.1-15.2
Помимо простой делеции, возможно появление кольцевой
хромосомы; мозаицизм по делеции; реципрокная
транслокация короткого плеча 5 хромосомы с другой
хромосомой с потерей критического участка.
34.
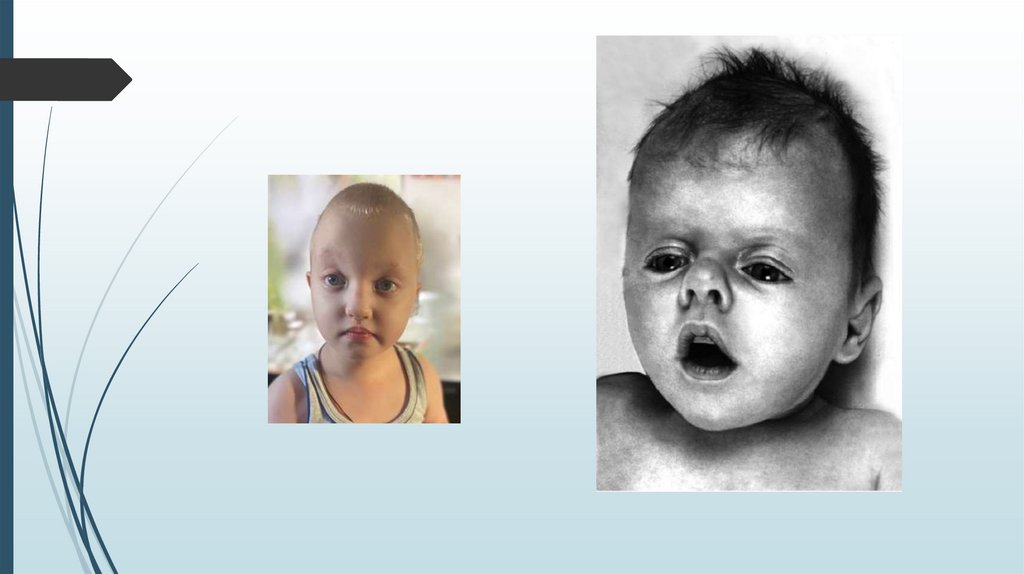
Синдром кошачьего крикаСимптомы
Необычный плач, напоминающий требовательное
мяуканье кошки. Обусловлен сужением гортани,
мягкостью хрящей, необычной складчатостью
слизистой оболочки.
Лунообразное лицо, микроцефалия, гипертелоризм,
микрогения, эпикант, высокое нёбо, плоская спинка
носа.
Ушные раковины деформированы и низко посажены.
Пороки сердца и других внутренних органов
Синдактилия стоп, клинодактилия 5 пальца кисти,
косолапость.
35.
Синдром кошачьего крикаСимптомы
С возрастом кошачий крик, лунообразное
лицо исчезают почти полностью.
Микроцефалия становится более отчетливой.
Заметнее психомоторное недоразвитие,
косоглазие.
Продолжительность жизни зависит от тяжести
ВПР, особенно сердца. Большинство больных
умирают в первые годы, около 10% достигают
10-летнего возраста. Имеются единичные
описания больных в возрасте 50 лет и старше.
36.
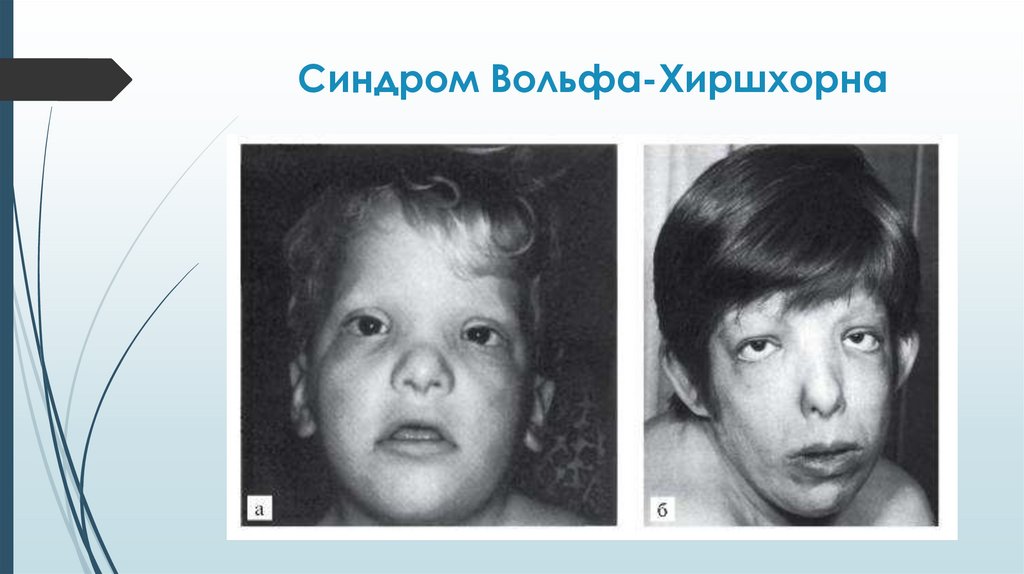
Синдром Вольфа-ХиршхорнаДелеция сегмента
короткого плеча
хромосомы 4 (4p-).
Частота 1:100 000.
37.
Синдром Вольфа-ХиршхорнаЦитогенетика синдрома характерна:
80% случаев у пробанда выявляется делеция
части короткого плеча 4р-, а у родителей
кариотипы нормальные.
Остальные случаи вызваны транслокациями или
кольцевыми хромосомами, но всегда при
этом отмечается потеря фрагмента 4р16.
38.
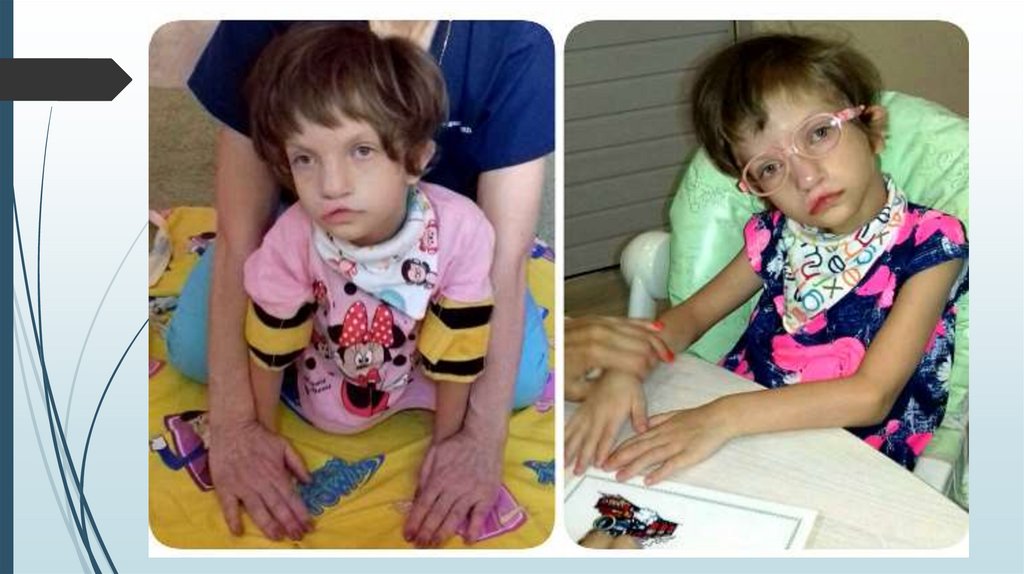
Синдром Вольфа-ХиршхорнаСимптомы
Многочисленные ВПР с последующей резкой
задержкой физического и психомоторного развития:
Уже внутриутробно отмечается гипоплазия плода.
Средняя масса тела детей при рождении 2000 г.
Микроцефалия, клювовидный нос, гипертелоризм,
эпикант, аномальные ушные раковины, расщелина
верхней губы и нёба, аномалии глазных яблок,
маленький рот, крипторхизм, гипоспадия,
деформация стоп и др.
ВПР внутренних органов – сердца, почек, ЖКТ – более
чем у 50% больных.
39.
40.
У большинствапациентов (50-100%)
возникает
эпилептический
синдром – судороги и
абсансы
41.
42.
Синдром Вольфа-Хиршхорна43.
Синдром Вольфа-ХиршхорнаЖизнеспособность детей
резко снижена. Большинство
умирает в возрасте до 1 года.
Описан один больной в
возрасте 25 лет.
44.
45.
46.
Анеуплоидиипо половым
хромосомам
47.
Синдром КлайнфельтераПолисомии по половым хромосомам. В
кариотипе не менее двух Х-хромосом и
не менее одной У-хромосомы.
Наиболее часто встречающийся и
типичный кариотип – 47, ХХУ. Встречается
в полном и мозаичном варианте с
частотой 1:750 новорожденных
мальчиков.
48.
49.
Синдром КлайнфельтераДо периода полового созревания
мальчики развиваются нормально, лишь
с небольшим отставанием в
психическом развитии.
Лишняя Х-хромосома проявляется в
период полового созревания в виде
недоразвития яичек и вторичных мужских
половых признаков.
50.
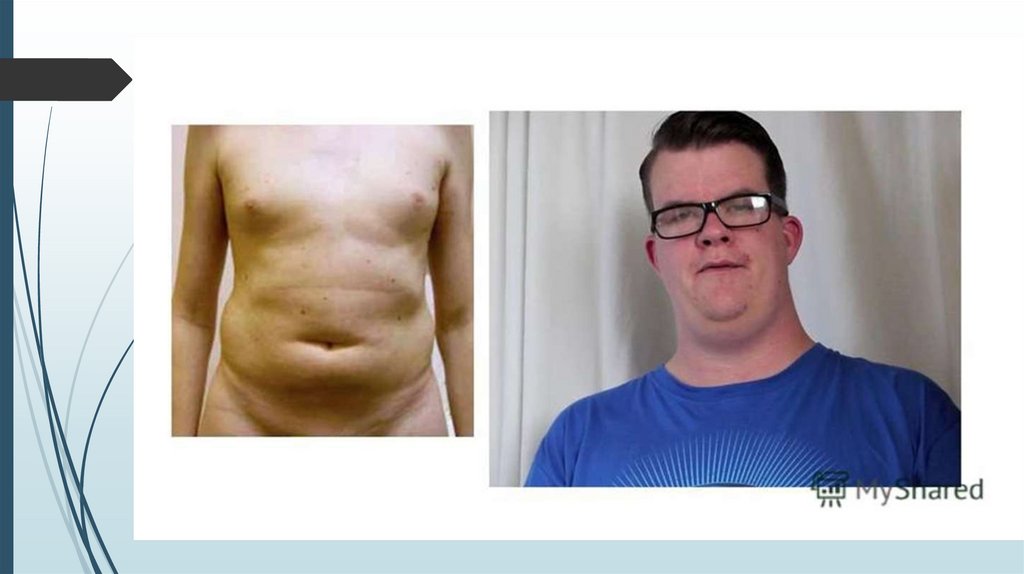
Синдром КлайнфельтераВысокий рост (максимальная прибавка роста в
5-8 лет), длинные конечности, высокая талия,
женский тип телосложения
Гинекомастия, слабое оволосение лица,
подмышечных впадин и лобка
Яички уменьшены. На гистологии дегенерация
эпителия и гиалиноз семенных канатиков.
Бесплодие (олигоспермия, азооспермия)
51.
Синдром КлайнфельтераТретья по распространенности эндокринная
патология у мужчин (после сахарного диабета и
гиперфункции щитовидной железы.
Наиболее частая причина нарушения
репродуктивной функции у мужчин.
Около половины случаев остаются
недиагностированными.
Причина – нарушение расхождения хромосом на
ранних стадиях формирования яйцеклеток и
сперматозоидов. Нарушение образования
яйцеклеток встречается в три раза чаще. Влияние
возраста родителей не выявлено.
52.
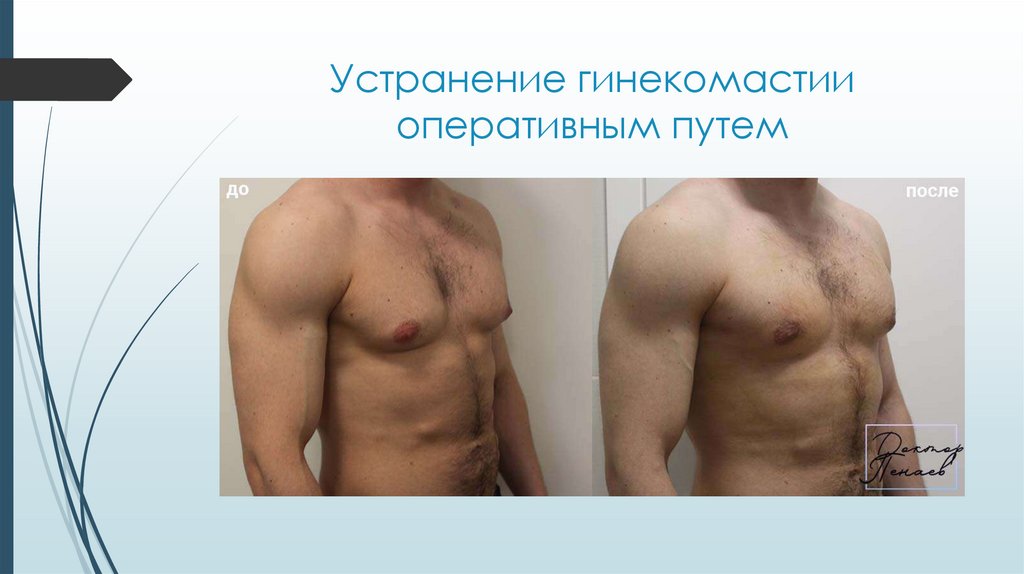
Устранение гинекомастииоперативным путем
53.
54.
Андрогены влияют на обмен веществ,поэтому больные синдромом
Клайнфельтера склонны к ожирению и
сахарному диабету 2 типа.
Статистически доказана
предрасположенность к аутоиммунным
заболеваниям (ревматоидный артрит,
системная красная волчанка)
55.
56.
Ителлектуальные особенностиКоэффициент интеллекта варьирует от
значений ниже среднего до высокого уровня.
Во всех случаях отмечается диспропорция
между уровнем интеллекта и вербальными
способностями: трудности при восприятии
больших объемов материала на слух, а
также при построении больших
грамматических фраз.
При добавлении каждой лишней хромосомы
коэффициент интеллекта снижается на 15 ед
57.
Психологические особенностиСкромные, робкие люди с
заниженной самооценкой и
повышенной чувствительностью
Есть данные, свидетельствующие о
склонности пациентов к
гомосексуализму, алкоголизму и
наркомании.
58.
Диагностика и лечениеПри подозрении на синдром Клайнфельтера
проводят анализ крови на определение уровня
андрогенов.
Точный диагноз ставят после определения кариотипа.
Пожизненная заместительная терапия препаратами
мужских половых гормонов позволяет улучшить
внешний вид, общее самочувствие, вернуть
способность к нормальной половой жизни,
предупреждает остеопороз и мышечную слабость.
В юном возрасте лечение надо начинать сразу после
постановки диагноза, что позволяет предупредить
гинекомастию.
59.
ПрогнозПри легком течении заболевания синдром
Клайнфельтера выявляют только при обращении
пациента по поводу бесплодия. Прогноз
относительно способности к оплодотворению
крайне неблагоприятный.
В последнее время появились сведения о появлении у
больных с синдромом Клайнфельтера здоровых
детей, рожденных с помощью технологии ЭКО. У
больных берут биоптат яичек, выделяют оттуда
зародышевые клетки, проводят оплодотворение in
vitro и подсаживают эмбрион в полость матки.
60.
Синдром дисомиипо У-хромосоме 47, ХУУ
Встречается с частотой 1:1000 новорожденных
мальчиков
Рост выше среднего, умственно развиты, не
дисморфичны
Заметных отклонений ни в половом развитии, ни в
гормональном статусе, ни в плодовитости у
большинства индивидов нет
Повышенного риска иметь хромосомно
аномальных детей нет
61.
62.
Синдром дисомии по У-хромосоме47, ХУУ
Почти для половины таких мальчиков требуется
дополнительная педагогическая коррекция в
связи с задержкой речевого развития,
затруднений в чтении и произношении.
IQ в среднем ниже на 10-15 пунктов.
Поведенческие проблемы: дефицит внимания,
гиперактивность и импульсивность, но без
выраженной агрессии или
психопатологического поведения.
63.
Синдром трисомии Х47, ХХХ
Среди новорожденных девочек частота
1:1000.
Имеют обычно нормальное физическое и
психическое развитие. Обычно выявляются
случайно при обследовании.
Нет отклонений в половом развитии,
фертильность в норме. При этом риск
хромосомных нарушений у потомства и
возникновения спонтанных абортов повышен.
64.
Синдром трисомии Х47, ХХХ
Интеллектуальное развитие в норме или на нижней
границе нормы.
У некоторых женщин с трипло-Х есть нарушения
репродуктивной функции (вторичная аменорея,
дисменорея, ранняя менопауза).
Редко встречаются варианты количества Х-хромосом
более 3. в этом случае нарастают отклонения от
нормы: в умственном развитии, черепно-лицевые
дисморфии, аномалии зубов, скелета и половых
органов. Однако женщины даже с тетрасомией могут
иметь потомство, повышен риск родить девочку с
трипло-Х или мальчика с синдромом Клайнфельтера.
65.
Синдром Шерешевского-ТернераЕдинственная форма моносомии у
живорожденных. Не менее 90% зачатий с
кариотипом 45, Х абортируется спонтанно.
Моносомия Х составляет 15-20% среди
всех аномальных кариотипов абортусов.
С возрастом матери не связан.
Частота 1:2000-1:5000 новорожденных
девочек.
66.
Синдром Шерешевского-ТернераЦитогенетика разнообразна:
Истинная моносомия 45,Х – 50-60%
Делеции короткого или длинного плеча
Х-хромосомы 46, Х, Хр- или 46, Х, Хq Изохромосомы 46, Х, (Хq)или 46, X, (Xp)
Кольцевые хромосомы 46, Х, R(X)
Различные варианты мозаицизма –
30-40%
67.
Синдром Шерешевского-ТернераСимптомы
Три основных направления:
1.Гипогонадизм, недоразвитие
половых органов и вторичных
половых признаков.
2.Врожденные пороки развития
3.Низкий рост
68.
Синдром Шерешевского-ТернераСимптомы
Половая система: отсутствие гонад (агенезия),
гипоплазия матки и маточных труб, первичная
аменорея, скудное оволосение лобка и
подмышечных впадин, недоразвитие молочных
желёз, недостаток эстрогенов, избыток
гонадотропинов гипофиза.
До 25% детей имеют врождённые пороки
развития сердца и почек.
69.
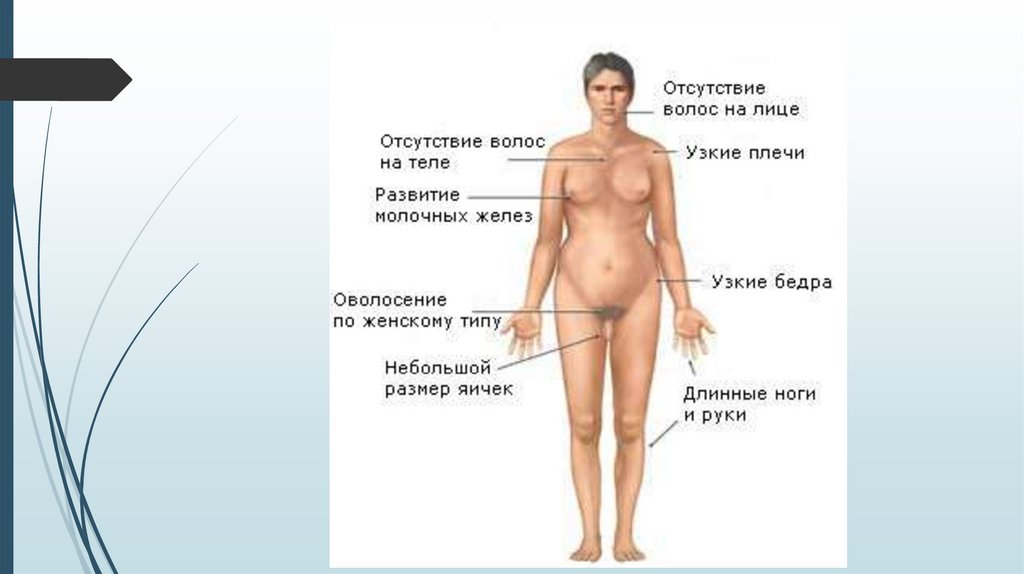
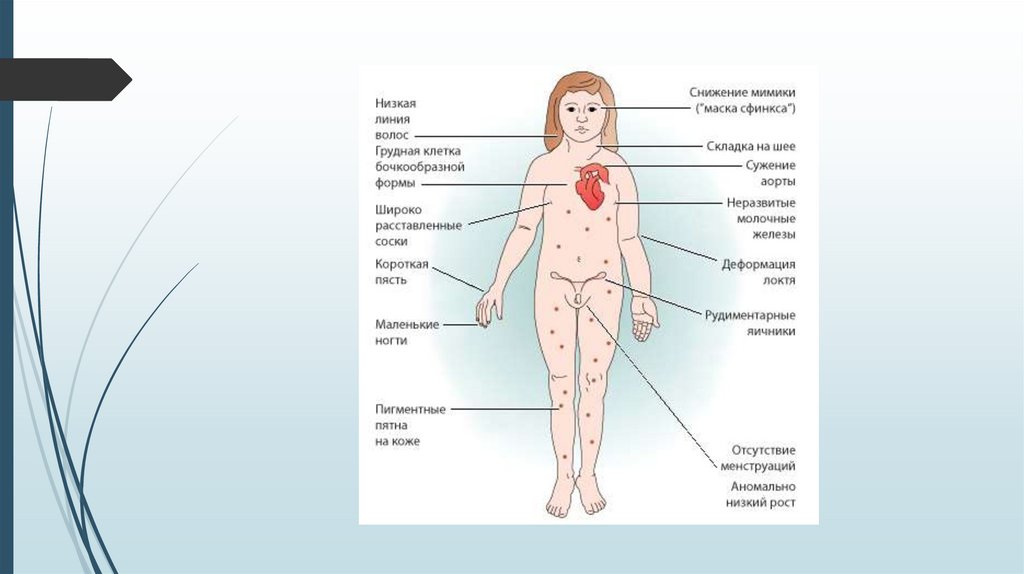
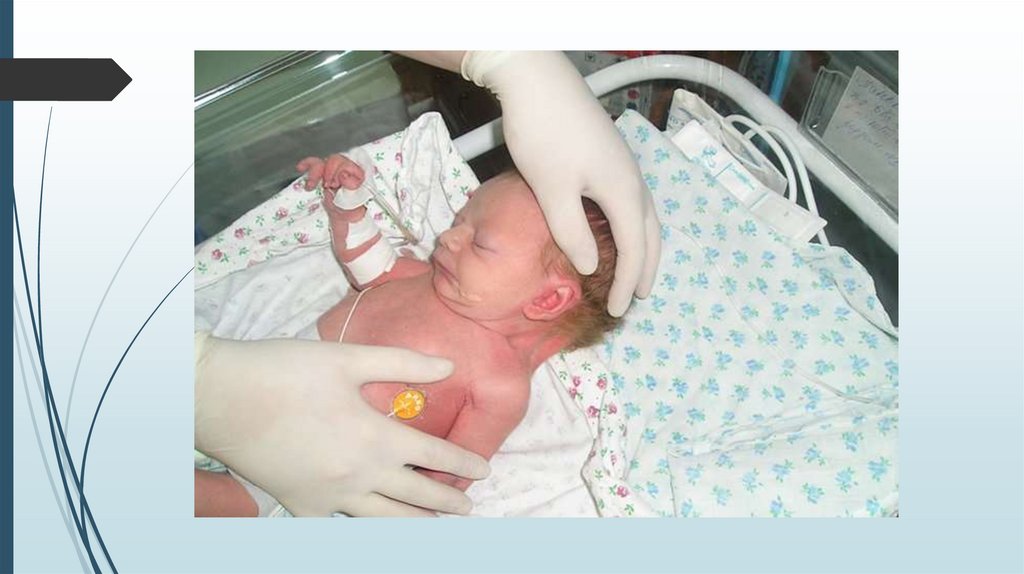
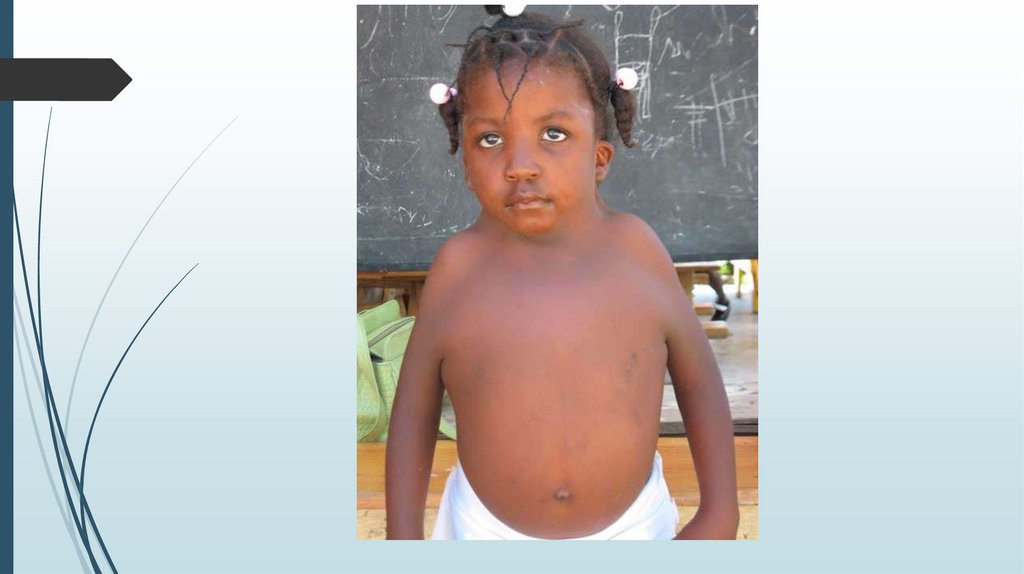
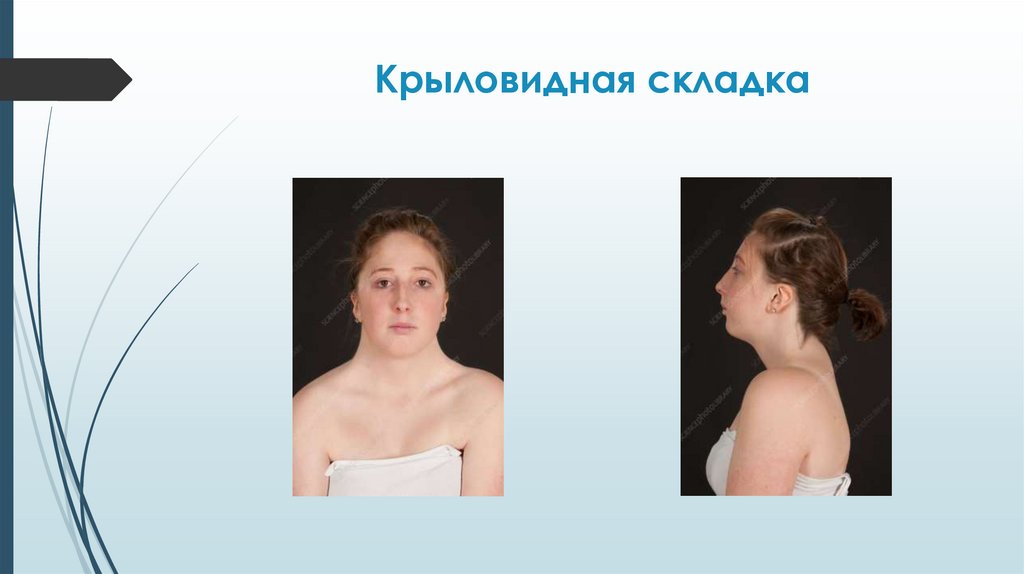
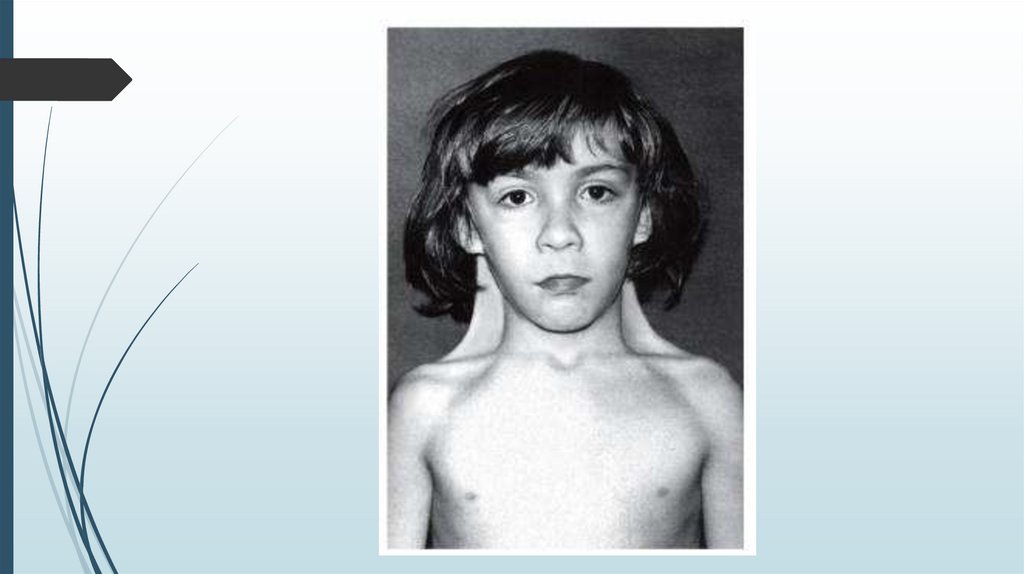
Синдром Шерешевского-ТернераВнешний вид
Короткая шея с избытком кожи и крыловидными складками.
Лимфатический отёк стоп, голеней, кистей рук и предплечий.
В школьном и подростковом возрасте отставание в росте, в
развитии вторичных половых признаков.
У взрослых нарушения скелета, черепно-лицевые
дисморфии, вальгусная девиация коленных и локтевых
суставов, короткие кисти, остеопороз, бочкообразная
грудная клетка, низкий рост волос на шее, птоз, эпикант,
низкое расположение ушных раковин. Рост взрослых девушек
на 20-30 см ниже среднего.
Мозаичные формы имеют более слабые проявления.
70.
Синдром Шерешевского-ТернераИнтеллект, как правило, сохранен; в редких
случаях отмечается олигофрения. Среди
сопутствующих заболеваний у лиц с синдромом
Шерешевского-Тёрнера обычно
выявляются гипотиреоз, витилиго,
множественные пигментные невусы,
алопеция, гипертрихоз, сахарный диабет 1-го и 2го типа, целиакия, ожирение, ИБС. У больных с
синдромом Шерешевского-Тёрнера значительно
повышен риск развития рака толстой кишки.
71.
ЛечениеВ раннем детстве лечение неспецифично - массаж, ЛФК,
витамины, полноценное питание, охранительный режим.
С целью увеличения конечного роста назначается рекомбинантный
гормон роста (соматотропин) в виде ежедневных подкожных
инъекций до достижения пациенткой костного возраста 15 лет и
уменьшения скорости роста до 2 см в год. В большинстве случаев
ростостимулирующая терапия помогает больным вырасти до 150–
155 см.
Для имитации нормального полового созревания с 13-14 лет
назначается заместительная терапия эстрогенами, а через 12–18
месяцев циклическая терапия эстроген-прогестагеновыми
оральными контрацептивами. Заместительная гормонотерапия
проводится до возраста естественной менопаузы у здоровых
женщин (примерно до 50 лет).
При ВПР хирургическая коррекция. Устранение крыловидных
складок шеи проводится методами пластической хирургии.
72.
73.
74.
75.
Крыловидная складка76.
77.
Спасибоза внимание!