
















 medicine
medicineSimilar presentations:





")



 (амстердамская карликовость)")
Синдром Кабуки
1.
С.Д. Асфендияров атындағы ҚазақҰлттық Медицина Университеті
Казахский Национальный
Медицинский Университет им. С.Д.
Асфендиярова
Тема: Синдром Кабуки
Подготовила: Қайырбаева А
Проверила: Гульнар А
2.
План:• Введение
• Этиология
• Патогенез
• Клиника
• Диагностика
• Лечение
• Литература
3.
• СиндромКабуки
–
это
редко
встречающееся моногенное заболевание,
характеризующееся
своеобразными
фенотипическими
признаками
и
интеллектуальной
дефицитарностью.
Больные имеют лицевые особенности
(миндалевидный разрез глаз, страбизм,
арочные брови, широкую переносицу, низко
посаженные оттопыренные уши и др.),
скелетные аномалии, пороки внутренних
органов, олигофрению.
4.
ЭтиологияПатология имеет гетерогенную этиологию.
Известно, что синдром является врожденным.
Большая часть диагностированных случаев
заболевания носит спорадический характер и
развивается вследствие мутаций, возникающих
«де
ново».
Факторы,
влияющие
на
изменчивость, не определены. В 2010-2012 г.г.
изучены две наиболее часто регистрируемые
наследственные формы синдрома, в основе
которых лежат мутации генов:
5.
• KMT2D(MLL2). Ген кодирует лизинспецифическую
метилтрансферазу
2D,
расположен в локусе 12q13.12. Мутация
вызывает первый тип заболевания (KS1) с
аутосомно-доминантным
наследованием.
Обнаруживается у 70% пациентов.
• KDM6A. Ген расположен на участке Xp11.3,
кодирует
лизин-специфичную
6А
деметилазу, выступает кофактором KMT2.
Генный дефект связан с развитием второго
типа
синдрома
(KS2),
имеющего
наследование, сцепленное с Х-хромосомой.
Составляет около 5% случаев патологии.
6.
ПатогенезВвиду неполной изученности причин синдрома Кабуки патологические
механизмы также остаются недостаточно ясными. Наиболее освещены
роль и влияние мутаций в генах KMT2D и KDM6A. Оба гена выступают
эпигенетическими регуляторами модификации гистонов (ядерных
белков) и играют важную роль в процессе экспрессии генов. Дефекты
кодируемых ими ферментов изменяют метилирование лизина в
гистоне H3, что приводит к развитию гетерогенных врожденных
аномалий
(черепно-лицевых,
скелетных,
висцеральных),
неврологических и эндокринных нарушений. Также у многих пациентов
с
синдромом
Кабуки
отмечаются
иммунные
нарушения,
проявляющиеся как снижением противоинфекционной защиты, так и
аутоиммунными расстройствами.
7.
Клиническая картинаКлиническая
картина
характеризуется
множественными
дефектами, затрагивающими различные анатомические системы:
лицевой скелет, опорно-двигательный аппарат, кожные покровы,
сердечно-сосудистую систему, ЖКТ, мочеполовые органы,
анализаторы
и
пр.
Также
выявляются
нарушения
неврологического, эндокринного, иммунного статуса различной
степени выраженности. Наиболее патогномоничны для синдрома
лицевой дисморфизм, который делает лица пациентов, похожими
на маску актеров театра кабуки, низкорослость и снижение
интеллекта.
8.
Лицевые аномалии включают микроцефалию,высоко поднятые (аркообразные) брови,
гипертелоризм, антимонголоидную глазную
щель, эктропион нижнего века. Дополняют
портрет больного с синдромом Кабуки низко
расположенные
торчащие
и
деформированные
уши,
широкий
приплюснутый нос, маленький рот с тонкими
губами. Реже отмечается голубоватая окраска
склер, небные расщелины, заячья губа,
микрогнатия, аномалии зубов. Своеобразное
выражение лица заметно уже в раннем
детстве, с годами же черты приобретают еще
более грубый и гротескный вид.
9.
ОсложненияПотенциальные
осложнения
тесно
связаны
с
теми
мультисистемными поражениям, которые имеются у ребенка. У
новорожденных возможны серьезные нарушения сосания и
глотания, требующие установки желудочного зонда или
гастростомы. Также в младенчестве отмечаются необъяснимые
эпизоды
гипогликемии,
угрожающие
переходом
в
гипогликемическую кому. У больных с синдромом Кабуки имеется
повышенный риск развития лимфолейкоза и неходжкинских
лимфом.
10.
ДиагностикаДиагноз обычно подозревается в первые месяцы после рождения, когда
родители отмечают изменения во внешности малыша, отличающие его от
других детей. Но иногда проявления могут выявляться в позднем возрасте.
Диагноз в основном устанавливается на основании фенотипичеких маркеров;
в крупных медицинских центрах доступно генетическое тестирование.
Первичная диагностика проводится генетиком, однако пациенты с синдромом
Кабуки требуют обследования и курации целым рядом специалистов:
педиатром, кардиологом, неврологом, ортопедом, урологом, иммунологом,
ортодонтом и т. д.
11.
Этапы обследования включают:• Портретную диагностику. Ядро признаков составляют лицевые стигмы,
отставание в росте, скелетные аномалии. Кроме высокочастотных
проявлений учитываются и редко встречающиеся атипичные признаки,
известные по описаниям отдельных клинических наблюдений (витилиго,
множественные гемангиомы, гипоспадия).
• Генетические
исследования. Выполняются в специализированных
лабораторных центрах. Позволяют изучить мутационный статус генов MLL2
и KDM6A. Для проведения высокоточной диагностики используются методы
флюоресцентной (FISH) и сравнительной геномной гибридизации на
микрочипах (aCGH). Обязательно обследование родителей на наличие
аналогичных мутаций.
12.
• Лабораторнуюдиагностику.
Гемограмма
характеризуется
тромбоцитопенией, лейкопенией, нейтропенией. Для обнаружения
сопутствующих нарушений проводятся гормональные и иммунологические
анализы. Исследуется гликемический профиль, уровень гормонов
щитовидной железы и гипофиза, иммуноглобулинов, ЦИК.
• Инструментальное обследование. Осуществляется с целью выявления
сопутствующих органных нарушений. Первоочередная диагностика
включает ЭхоКГ, УЗИ органов брюшной полости и почек, рентгенографию
костей и позвоночника. При эписиндроме выполняется видео-ЭЭГмониторинг.
Дополнительно
проводится
офтальмологическое
обследование, аудиометрия.
• При высоких рисках рождения ребенка с генетическими аномалиями
назначается инвазивная пренатальная диагностика с последующим
полногеномным секвенированием.
13.
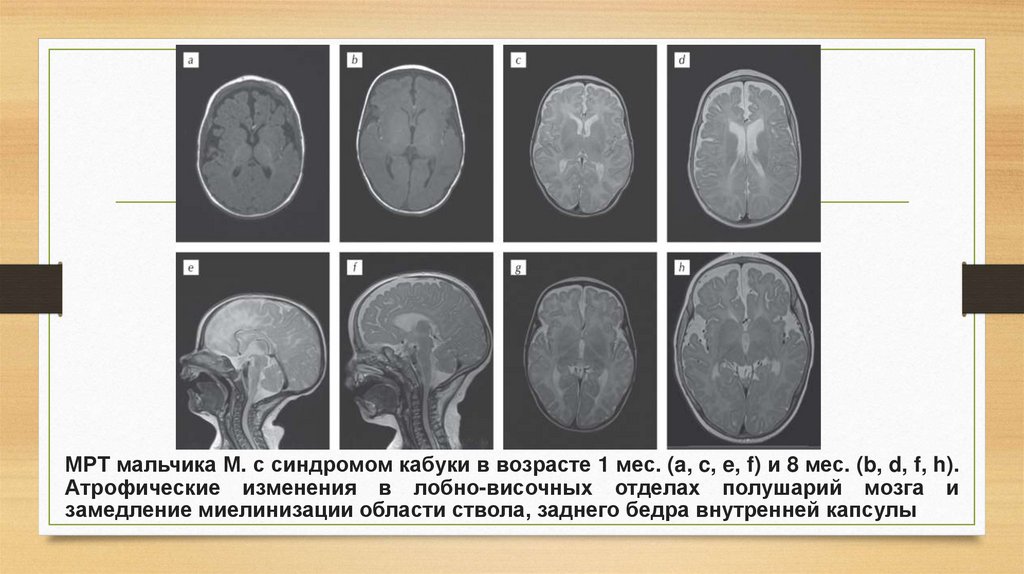
МРТ мальчика М. с синдромом кабуки в возрасте 1 мес. (a, c, e, f) и 8 мес. (b, d, f, h).Атрофические изменения в лобно-височных отделах полушарий мозга и
замедление миелинизации области ствола, заднего бедра внутренней капсулы
14.
Дифференциальная диагностикаДифференциальная
диагностика
осуществляется
с
другими
генетическими синдромами, сопровождающимися множественными
нарушениями развития:
• Ди Джорджи,
• Ван-дер-Вуда,
• Мельника-Фрейзера,
• Элерса-Данло,
• Чарджа и другими.
15.
ЛечениеВвиду генетической детерминированности заболевания полное
излечение невозможно. Стандарты терапии синдрома не
регламентированы, на практике применяется симптоматический
подход к устранению тех нарушений, которые мешают развитию
ребенка. Хирургическая патология (грыжи, ВПС, расщелины ЧЛО,
атрезия анального канала, крипторхизм и т.п.) устраняются в
ходе оперативного лечения.
16.
Сопутствующиеназначением:
аутоиммунные
расстройства
корригируются
• глюкокортикоидов,
• иммуносупрессоров,
• моноклональных антител.
Рецидивирующие
инфекционные
процессы
диктуют
необходимость
применения
антибактериальных
средств,
внутривенного введения человеческого иммуноглобулина. При
выявлении диабета 1-го типа решается вопрос о назначении
инсулинотерапии.
17.
ПрофилактикаДети, страдающие синдромом Кабуки, нуждаются в постоянном уходе
и наблюдении, социальной адаптации, инклюзивном образовании.
Семье, воспитывающей такого ребенка, требуется психологическая
поддержка. В связи с большим количеством сложных дефектов
качество и продолжительность жизни пациентов снижены. Выяснить
вероятность наследования синдрома Кабуки ребенком при наличии
семейных
случаев
патологии
позволяет
генетическое
консультирование.
18.
Литература• Русский журнал детской неврологии (том VI выпуск 3 2011)
Интернет-сеть
• https://www.krasotaimedicina.ru/diseases/genetic/Kabuki-syndrome
• https://fnkc.ru/hemoncim/art/16-4/2017-16-4-75-83.pdf